
Fluorination reaction of UO3 and electrochemical preparation of UO2
2022-07-09 02:14RugengLiuYangyangMengWenjingJiWeiHanMeiLiYangSun
Chinese Chemical Letters 2022年7期
Rugeng Liu, Yangyang Meng, Wenjing Ji, Wei Han, Mei Li, Yang Sun
Key Laboratory of Superlight Materials and Surface Technology, Ministry of Education, College of Material Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China
ABSTRACT In this work, a technique was proposed to prepare UO2 from UO3 by the two processes of fluorination reaction of UO3 with NH4HF2 and electrochemical reduction of UO2+2 for the recycle uranium.The feasibility of fluorination reaction was firstly confirmed using thermodynamic calculation; then, the products were analyzed using XRD, Raman and fluorescence to be UO2F2.The fluorination mechanism was inferred to be UO3(s)+NH4HF2 →(NH4)3UO2F5→NH4(UO2)2F5 →UO2F2.The redox behavior of UO2+2 on W electrode was investigated by cyclic voltammetry and square wave voltammetry, which indicated that UO2+2 was reduced to UO2 via a two-step single electron transfer with diffusion-controlled.The diffusion coefficient of UO2+2 was calculated to be 6.22×10-5 cm2/s.The disproportionation reaction of UO+2 was observed, and the relationship between the disproportionation reaction and scan rate was discussed.Moreover, the electrochemical fabrication of UO2 was conducted by electrolysis at -0.8 V, and the product was analyzed by XRD, SEM and EDS to be UO2.ICP-AES results showed that the extraction efficiency of UO2 could reach 98.53%.
Keywords:UO3 Fluorination reaction NH4HF2 Electrochemical formation UO2
The reprocessing of spent fuel is one of the critical steps in the nuclear fuel cycle.In recent decades, extensive studies have been carried out on dry reprocessing using molten salt as a medium[1–3].Thereinto, the electrolytic refining process developed by the United States and oxide-electrowinning reprocessing developed by Russia as are considered as the prominent candidate technologies for advanced nuclear fuel reprocessing in the future [4,5].For MOX spent fuel, removing the cladding material is an important step in head-end process of spent fuel reprocessing.Recently, high temperature oxidation technology was adopted to separate spent fuel from cladding material by high temperature calcination UO2to form a volume expansion force to destroy the cladding material,which was proposed by Idaho national laboratory (TNL) and Korea atomic energy research institute (KAERI).During the process, UO2is oxidized to U3O8and UO3[6].In the oxide-electrowinning process, uranium and fission products in the oxide fuel are firstly chlorinated and dissolved into the molten salt as oxychloride (uranyl ion) and chloride ions by flowing chlorine gas, then uranyl ion is reduced to form the raw material of MOX fuel (UO2) on the cathode in molten chlorides.However, the spontaneity of the chlorination reaction is unfavorable, and hence, it is necessary to add reducing agents, for instance, carbon and carbon monoxide into the melts [7].Thus, many investigators explored the chlorination reaction of uranium oxide (UO2and U3O8) using ZrCl4[8], CCl4[9,10], NH4Cl [11].Sakamuraet al.[8] studied UO2was chlorinated to form UCl4by ZrCl4in LiCl-KCl melt, however, the conversion rate was only 37%.Kitawakiet al.[9] explored the chlorination UO2and U3O8using CCl4through mechanical mixing method, but did not succeed.Jianget al.[10] chlorinated U3O8to UCl4using CCl4at high temperature, but CCl4would volatilize severely and the by-product of UCl5was formed.As we known, UCl5is easy to volatilize which causes the loss of uranium.Liuet al.[11] investigated the chlorination reaction of U3O8and NH4Cl in LiCl-KCl melt under air atmosphere.Their results showed that U3O8can be successfully chlorinated to.Waniet al.[12] studied the fluorination of UO2and U3O8with NH4HF2and found the formation of[NH4]4UF8and [NH4]3UO2F5compounds.These two compounds,on heating to about 673 K could form UF4and UO2F2·2H2O, respectively.
The electrochemical reduction ofhas been explored in molten chlorides and found thatcan be reduced to UO2by two-step single electron transfer [13–16].Schlechteretet al.[17] reported that the crystalline form of electrodeposited UO2in LiCl-KCl molten salt was affected by the presence of fluoride, and the presence of a small amount of fluoride would be conducive to the formation of cubic crystal UO2.Caligaraet al.[18] measured the diffusion coefficient ofin molten LiCl-KCl eutectic.
In order to explore the route of the formation of UO2using UO3as raw material, NH4HF2was chosen as fluoride reagent to make uranium oxide change tounder air atmosphere.Then,the electrochemical formation of UO2was investigated in LiCl-KCl molten salt by constant potential electrolysis for uranium extraction.The fluoride product and electrodeposition sample were checked by XRD, SEM-EDS.
Firstly, the possibility of the formation of UO2F2from UO3was estimated using the thermodynamic analysis of related fluorination reactions.UO3can react with NH4HF2or HF obtained from the decomposition of NH4HF2to form UO2F2, the following reactions may be involved as follows:
The standard Gibbs free energy changes(Δr)for these reactions at various temperatures were calculated shown in Fig.S1(Supporting information).It can be seen that(Δrof these reactions are less than zero, which means that all the reactions correlated with the formation of UO2F2are spontaneous at the temperature of 473–873 K.Moreover, the(Δr)becomes more and more negative with the increasing temperature, indicating that the increase of temperature can be beneficial to the fluorination reaction of UO3.
The fluorination reaction of UO3with NH4HF2was performed at 573 K for different times.The obtained products were characterized by XRD shown in Fig.1a.It can be seen from Fig.1a that after the fluorination reaction for 30 min, only (NH4)3UO2F5is observed except for the unreacted NH4HF2.No diffraction peak of UO3occurs, indicating that UO3is firstly converted to (NH4)3UO2F5.After 60 min, only NH4(UO2)2F5appears, which shows that (NH4)3UO2F5is completely converted to NH4(UO2)2F5.When the fluorination reaction time is extended to 120 min, the diffraction peak of UO2F2is observed.However, NH4(UO2)2F5is not completely decomposed,and its diffraction peaks can still be observed.However, after the reaction for 420 min, XRD result indicates there are only UO2F2,and NH4(UO2)2F5has been completely converted to UO2F2.
To further confirm the formation of UO2F2, fluorescence spectroscopy and Raman spectroscopy were used to analyze the product obtained from the fluorination of UO3for 180 min at 673 K.It can be seen from Fig.1b that peaks maxima appear at about 485, 495, 516, 538 and 566±0.5 nm, showing a five-finger shape,which is a typical characteristic peak of[19,20].Fig.1c shows the Raman spectrum of the product.Three obvious vibration peaks are observed at 181 cm-1, 443 cm-1and 915 cm-1, respectively.Among them, the strong band at 915 cm-1is ascribed to the symmetric U-O stretching vibration.The band at 181 cm-1corresponds to the U-O bending fundamental and the weaker band at 443 cm-1is related to the U-F lattice vibration which are consistent with those of UO2F2obtained by Armstronget al.[21].These results demonstrate the formation of UO2F2.
The reaction mechanism is expressed as follows:
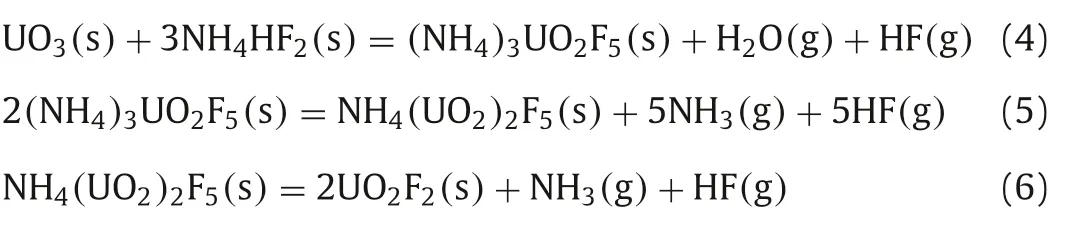
The products obtained under different fluorination times at 573 K were analyzed by SEM to observe the morphology changes.Fig.S2 (Supporting information) shows the SEM images of fluorination products obtained at different duration.It can be seen that UO3presents a spherical particle with size of about 1 μm.UO3was fluorinated for 30 min, according to the XRD result, (NH4)3UO2F5with size of about 3 μm is formed.A truncated octahedral composed of four hexagons and ten quadrilaterals is observed in Fig.S2b.After 60 min of the reaction, according to the XRD result, (NH4)3UO2F5is decomposed to NH4(UO2)2F5, and NH3and HF are released.Thus, a small number of pores are observed on the surface of the particles (Fig.S2c).When the reaction proceeds for 420 min, the product is UO2F2based on the XRD result.It can be observed from Fig.S2d that the formation of pores appears on the surface of the particles due to the release of NH3and HF, which indicates that large amounts of NH3and HF have been released.
Then the electrode reaction ofwas studied in LiCl-KCl melts on Mo electrode at 773 K using cyclic voltammetry (CV)shown in Fig.2a.In the black line, peaks C1and A1are attributed to the deposition and dissolution of metallic Li.Whilewas added into LiCl-KCl melts, the color of the molten salt change from colorless to yellow shown in the inset of Fig.2a, which is exactly the color ofdissolved in molten salt.In the red line, four new anodic peaks, A2, A2’, A3and A4, are observed at -2.21 V, -1.42 V,0.27 V and 0.54 V, respectively.Only three cathodic peaks, C2, C3and C4, are observed at -2.45 V, -0.31 V and 0.32 V, respectively.The redox couple C4/A4may be correlated with the soluble/soluble redox process of

The C3/A3couple pertains to the formation of UO2and its reoxidation.

The C2/A2couple is ascribed to the reduction of UO2to U metal and oxidation of U metal, and A2′ corresponds to the dissolution of U to U3+[22,23].
Fig.2b presents the CV curves of LiCl-KCl-(0.24 wt%)melts at various scan speeds.For the redox peaks of C3/A3, with the increase of the scan rate, the cathodic/anodic peak currents increase and the cathodic/anodic peak potentials have a significant negative/positive shift which shows that the Eq.9 is not a reversible process.However, for the redox peaks of C4/A4, with the increase of the scan rate, the cathodic peak current increases and anodic peak current decreases.It can be seen that when the scan rate is 0.04 V/s and 0.06 V/s, the oxidation peak A3is not observed.A4is related to theto disproportionate into UO2(monolayer)and it has a sharp shape and a large peak current.When the scan rate reaches 0.08 V/s, peak A3appears, and the peak current of A4decreases rapidly, the peak potential shifts negatively, and the peak become widens.When the scan rate increases from 0.1 V/s to 0.16 V/s, the peak current of A3continued to increase, the peak current of A4gradually decreases, and peak A4became wider.
This phenomenon may be caused by the disproportionation reaction of

At low scan rate, the measurement time is relatively long, the formedhas enough time to generateand UO2by the disproportionation Eq.9, and the concentration ofdecreases,which results in that the current peaks C3and A3related to the reduction and oxidation ofare not obvious.As the scan rate increases, the disproportionation reaction time is reduced, and the amount of UO2generated by the disproportionation reaction gradually decreases, the peak current of the oxidation peak A4gradually decrease and oxidation peak A3gradually appears.
The electrode area is 0.3768 cm2.Assuming that a layer of UO2was deposited on the electrode surface, the electric quantity of A4which was integrated of the peak area required by disproportionation reaction is at least 0.102 C.To obtain the electric quantity passed during oxidation reaction, the current and oxidation time related to the peak A4in Fig.2b was integrated, and then the relationship of quantity of electricity versus scan rate was plotted,as shown in Fig.2c.It can be seen that when the scan rate is relatively low, the electric quantity passing through the oxidation peak A4is close to that required to theoretically deposit a single layer of UO2.As the scan rate increases, the electric quantity passing through peak A4gradually decreases, which meant the amount of UO2deposited at A4gradually decreases.This is consistent with the analysis result in Fig.2b and further proves that the existence of oxidation peak A4is correlated with the formation of UO2by the disproportionation reaction of.
To further illustrating the electrochemical reactions correlated with peaks C3and C4, the square wave voltammetry (SWV) was used to measure the number of electron-transferred during the two electrochemical reactions.Fig.2d shows the SWV curve when reduction reaction ofoccurs on W electrode.In the measured potential range, three reduction peaks (C3, C3′ and C4) are observed and then the number of electrons transferred obtained by Eq.10.

where W1/2is the half-peak width; R is ideal gas constant; T is absolute temperature; F is Faraday constant; and n is the number of electrons transferred.
Since the appearance of peak C3’, which may be related to the nucleation of UO2[24], will interfere with the calculation result peak C3, the Gaussian fitting was performed to eliminate the influence.Using Eq.10, the number of electron transfer attributed to the peak C3is 1.09, close to 1.Thus, the reaction correlated with peak C3is the reduction reaction ofto UO2via a one-step involving one electron.
And the calculated number of electron-transferred from the reduction peak of C4is 0.92, close to 1, which means the reaction pertaining to peak C4is thatis reduced toin a one-step with the exchange of one-electron.Fig.2d shows the SWV curves of LiCl-KCl-UO2F2melts at various frequencies.As shown in Fig.2e,a pair of obvious redox peaks A4’/C4appears at around -0.40 V,and their shape is flat and symmetrical, which indicates that the redox reaction is a reversible reaction in a soluble/soluble system.Plotting the peak current density and the square root of the frequency, a good linear relationship indicates that the redox reaction is controlled by diffusion (Fig.2f).Thus, the diffusion coefficient ofcan be calculated using Eq.11 [25].
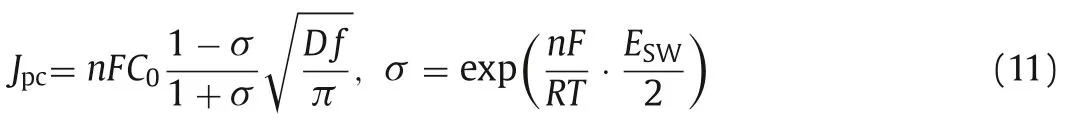
where C0is concentration ofin the molten salt(C0=9.804×10–5mol/cm3); D is diffusion coefficient (cm2/s);n is numbers of electrons transfer (n=1); f is frequency; ESWis amplitude of SWV (ESW=0.1 V).
The calculated diffusion coefficient ofat 773 K is 6.22×10–5cm2/s, which has the same order of magnitude with the literature (1.54×10–5cm2/s) obtained on platinum electrode at 858 K [18].
According to the electrochemical results, it is feasible for the formation of UO2by molten electrolysis.Thus, potentiostatic electrolysis was performed to form UO2at -0.8 V for 8 h.The change of current with time was recorded, as shown in Fig.3a.A strong current appears at the beginning (inset), which might be related to the formation and growth of UO2nuclei.Before 1500 s, the current fluctuated at -20 mA, meaning the formation of UO2.However,with the proceeding, the concentration ofdecreases, leading to the decrease of the absolute value of current.When the I-t curve became steady and close to background current about 2 mA,it could be considered that the run is completed.
The XRD spectra of the product gained shown in Fig.3b indicates the formation of UO2successfully and the color of the Mo plate changes from metallic luster to black, as shown in inset of Fig.3b.After stripping off the UO2from the electrode, the diffraction peaks in XRD spectra shown in Fig.3b became stronger and sharper, showing the deposited UO2has a high crystallinity.The morphology and element distribution were characterized, as shown in Figs.3c-f, the UO2is unevenly distributed on the electrode surface in a dendritic state.EDS analysis also proved the formation of UO2.Before and after the run, the concentration of U in the melts was analyzed by ICP-AES and the extraction efficiency was calculated using Eq.12.The calculated result indicated that after 8 h electrolysis, the extraction of UO2can reach 98.53%.

where η is extraction efficiency; C1and C2are the initial and final concentrations ofin the melts, respectively.
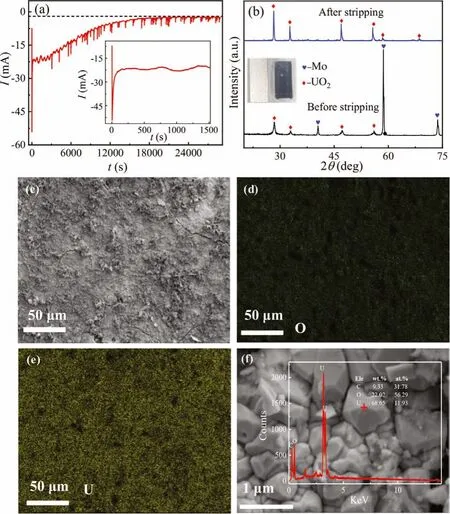
Fig.3.(a) I-t curve, (b) XRD patterns and (c) SEM-EDS images of electrolysis product obtained by potentiostatic electrolysis at -0.8 V on Mo plate at 773 K.
In order to electrochemically form UO2using UO3as raw material, the formation of UO2F2by the fluorination reaction of UO3and NH4HF2and its mechanism were firstly studied using XRD, Raman and fluorescence.The results revealed the formation of UO2F2, and the mechanism was inferred to be comprised of the following three process UO3(s)+ 3NH4HF2(s)→(NH4)3UO2F5→NH4(UO2)2F5(s)→2UO2F5(s).Then, the electrochemical formation of UO2was studied using CV and SWV, which illustrated UO2+2 could be reduced to UO2viaa two-step single electron exchange process with diffusion-controlled.The diffusion coefficient ofwas estimated to be 6.22×10-5cm2/s.The disproportionation reaction ofwas observed, and the relationship between the disproportionation reaction and scan rate was discussed.In addition, UO2was prepared employing potentiostatic electrolysis at -0.8 V for 8 h in LiCl-KCl-UO2F2melt.XRD and SEMEDS results indicated UO2with micron-sized octahedral structure was formed, and the extraction efficiency of UO2calculated by ICPAES could reach 98.53%.The feasibility of UO2prepared using UO3as raw material was proved.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The work was financially supported by the National Natural Science foundation of China (Nos.U2167215, 22076035, 21876034,11875116 and 21790373), and the Fundamental Research Funds for the Central Universities (No.3072021CFJ1001).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.069.
 Chinese Chemical Letters2022年7期
Chinese Chemical Letters2022年7期
- Chinese Chemical Letters的其它文章
- Professor Zhifang Chai: Scientific contributions and achievements
- Stable isotope labeling of nanomaterials for biosafety evaluation and drug development
- Emerging nanozymes for potentiating radiotherapy and radiation protection
- Recent progress of astatine-211 in endoradiotherapy: Great advances from fundamental properties to targeted radiopharmaceuticals
- Recent development in selective Tau tracers for PET imaging in the brain
- 64Cu radiolabeled nanomaterials for positron emission tomography(PET) imaging