
The development of molecular and nano actinide decorporation agents
2022-07-09 02:14XiaomeiWangCenShiJingwenGuanYemengChenYigongXuJuanDiwuShuaoWang
Chinese Chemical Letters 2022年7期
Xiaomei Wang, Cen Shi, Jingwen Guan, Yemeng Chen, Yigong Xu, Juan Diwu,Shuao Wang
State Key Laboratory of Radiation Medicine and Protection, School for Radiological and Interdisciplinary Sciences (RAD-X) and Collaborative Innovation
Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China
ABSTRACT Internal contamination of actinides has led to significant health hazards to the public and workers in the context of nuclear power plant accidents, uranium ore mining, and reprocessing of the used fuel.An effective sequestering agent that is able to remove accidentally incorporated actinides in vivo with low toxicity is always in urgent need.The molecular decorporation ligands have been the most widely researched agents for the past few decades, while preliminary studies of functionalized nanoparticles have shown their clear advantages in metal binding selectivity, toxicity, and oxidative stress alleviation.Herein,the state-of-the-art of those two types of decorporation agents is presented with special attention being paid on the correlation between the solution and solid-state chemistry of those agents with actinides and the corresponding decorporation efficacies.
Keywords:Actinides Chelator Binding selectivity Coordination mode Decorporation efficiency
1.Introduction
Nuclear safety has renewed a high level of worldwide concern for tragic lessons learned from Fukushima and Chernobyl nuclear power station accidents [1,2] as well as the occupational exposure of workers in the nuclear industry [3,4].In the nuclear accident scenario, actinides as nuclear materials and radioactive products of nuclear reactions may leak from the site and cause serious pollution to the environment, further inducing internal contamination to humanviathree most common induction routes (i.e., ingestion,inhalation, and wound-related absorption).Besides, workers facing risks of potential occupational exposures in the processes of fuel recycling, radioactive waste treatment, and uranium mining should also be taken care of [5,6].Due to the chemotoxicity and radiotoxicity of actinide ions, their uptake by human will cause severe health effects [7].For instance,239Pu is a fissionable material and is considered one of the most toxic radioactive substances, whose acute or long-term internal radiation and heavy metal toxicity will induce organ dysfunction or even cancers, such as lung cancer and osteosarcoma [7–11].The most plausible treatment of people with internal exposure of actinides is considered as the decorporation method, where a specific chelator is utilized to selectively bind actinides and form stable complexesin vivo, thereby preventing the deposition in target organs, accelerating the excretion, and consequently reducing the overall accumulation of actinides in tissues[12].
In the past few years, thanks to the continued efforts that the researchers have devoted, a large amount of chelators have been developed for the purpose of actinide decorporation [7].General rules apply in the design of an efficient chelating agent, such as high binding selectivity, low toxicity and oral activity.From the coordination chemistry perspective, actinide ions, as hard cations(strong Lewis acids), prefer to coordinate with hard anions(e.g., N,O, F), and the functional groups containing those atoms such as hydroxyl, carbonyl, and carboxyl moieties are preferred.More importantly, the coordination number and geometry of actinides vary a lot mainly based on valence states [13–16], so matched geometry and denticity (donor atom numbers) are the primary prerequisites to achieve strong actinide coordination ability with high selectivity.
Here in this review, the fundamental knowledge of actinides,such asin vivodistribution, stable valence states, speciation and/or reactions with the relevant biological molecules is provided firstly.Then a list of the selected actinide decorporation agents to date is introduced, together with the coordination mode analysis, the complexing ability of ligands and the adsorption behavior of nanochelators, and their decorporation efficiency for the targeted actinide ions (i.e., Th, U, Np, Pu, and Am).Finally, the correlation between binding affinity and selectivity, coordination mode, and removal efficiency of actinide decorporation agents is briefly discussed.
2.Actinide distribution and speciation in vivo
Three main pathways for the uptake of actinides are inhalation,ingestion, and skin exposure (Fig.1).Based on current reports of human contamination in accidents, inhalation is the most common one among the three routes, followed by skin contamination, especially on wounds [10,13,15].In the case of environmental contamination of radionuclides, the ingestion of contaminated food and water poses a health threat to civilians [17–19].Generally, as illustrated in Fig.1, the chemical forms of actinides play an important role in their distributionviadifferent pathways.
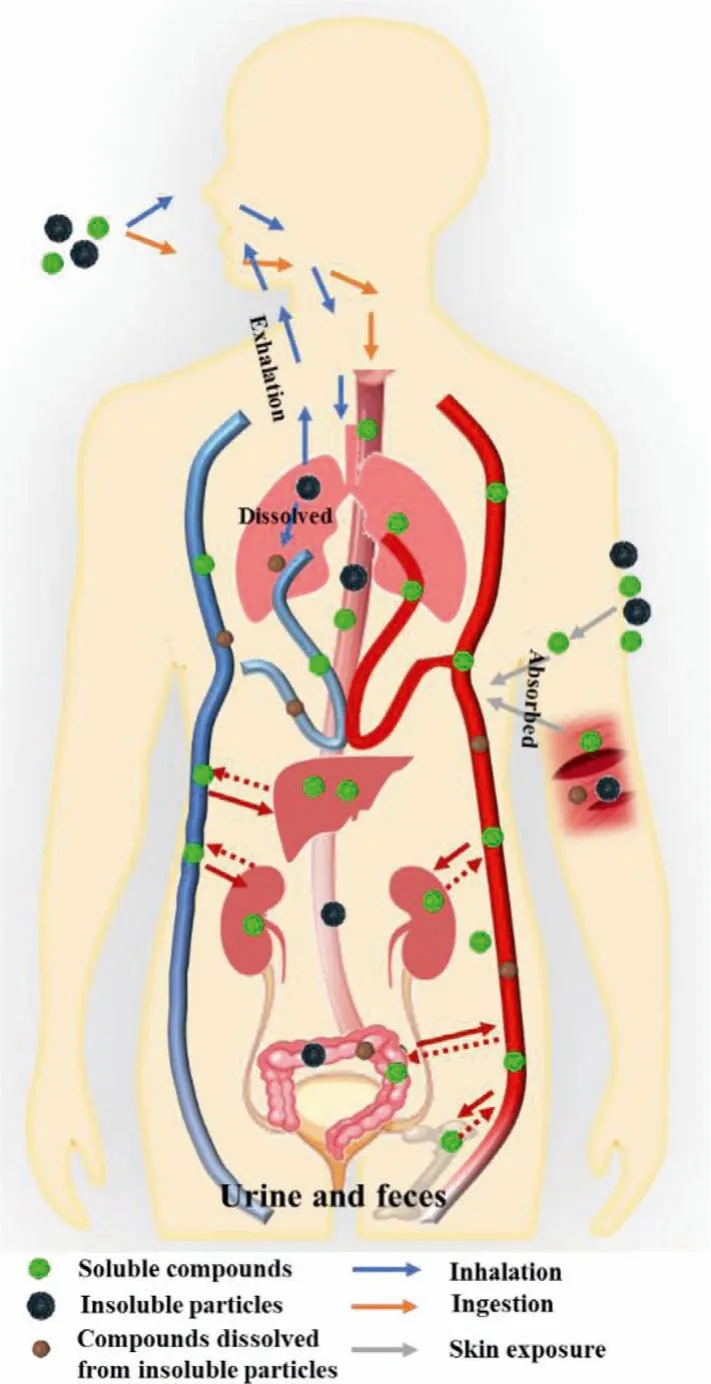
Fig.1.Possible pathways of actinides.
For inhalation, the pulmonary clearance rate of inhaled actinide particles varies according to their chemical forms and particle sizes.Mucociliary action can deliver large particles from the respiratory system to the pharynx, where these particles are swallowed and finally eliminated in the feces.The actinide compounds deposited in the pulmonary tissues, especially the pulmonary lymph nodes, are difficult to be eliminated from the body [10,11].Insoluble actinide compounds, such as oxides (AnOx), present a long retention time, resulting in lung injury [21].In contrast, soluble actinide compounds, such as nitrates, carbonates, and chlorides are dissolved in the pulmonary alveoli and rapidly enter the blood.The more soluble actinide compounds are, the easier they are absorbed into the bloodstream, deposited in tissues, and excreted [20].For ingestion, insoluble actinide particles are difficult to be absorbed by intestinal epithelial cells and eliminated in the feces.In contrast, soluble ones are easily absorbed into the bloodstream [22].For skin exposure, insoluble actinide compounds mainly remain in the contamination site, while some of them may be slowly dissolved by sweat or fluids (wounded sites) and absorbed through the transdermal pathway [23,24].
The speciation of actinide ions in blood vary a lot primarily due to their differences in stable oxidation states and ionic radius[25,26].A unique feature of penta- and hexavalent actinide ions is their presence as the actinyl unit consisting of a tri-atomic linear unit of [O=An=O]+,2+.The coordination number of An(IV) and An(III) is generally in the range from 7 to 9, with 8 being the most common one.In contrast, the actinyl units tend to bind with 4 to 6 donor atoms equatorially.A general order of the complexation stability and hydrolysis tendency of actinide ions is found as An4+>AnO22+≈An3+>AnO2+.Due to the high charge of tetravalent actinide ions, they are easily hydrolyzed to form colloidal particles.Besides, An(IV) ions readily react with transferrin (Tf) and form stable An(IV)-Tf complexes.Th(IV) is the largest in ionic radius and IV is its only stable oxidation state [25,26].Approximately 90% of Th(IV) forms the Th(IV)-Tf complex and is then eliminated from the blood within one day [9].At least 80% of thorium is deposited in tissues (mainly in liver and bone) [26,27].In contrast,plutonium exhibits active redox chemistry with possible oxidation states of III, IV, V and VI, and IV is the most stable oxidation statein vivo viaa series of disproportionation reactions [16,28,29].Compared to Th(IV), Pu(IV) is significantly smaller in ionic radius and therefore higher in the surface charge.Moreover, Pu(IV) resembles Fe(III) in the biological transport and distribution properties on account of their similar charge to ionic radius ratio (4.6 and 4.3 e/, respectively) [30,31].As a result, transferrin gives higher affinity toward Pu(IV) and forms 100% Pu(IV)-Tf complexes [30].Then, 90% of the total plutonium uptake is eliminated in plasma and accumulates in the liver (~30%) and bone (~50%) within 24 h[26,29,32].
IV and VI oxidation states are considered to be stable for uranium [25,33].However, VI is the most stable state of uranium.When taken up into the body, other forms of uranium, including U(IV), are eventually oxidized to hexavalent uranyl ions (UO22+,U(VI)) [7,33].In the bloodstream, approximately 50% uranium exists in the form of carbonate, 30% of uranium reacts with transferrin, and the remaining 20% of uranium accumulates in red cells[34,35].Uranium excretes quickly from the blood, since only 9.1%and 1.6% of U(VI) injected remain in the blood 10 min and 1 h later,respectively.Approximately 2/3 of the injected uranium is excretedviaurine.The rest eventually deposits in the kidneys and bones as hexavalent uranyl ions (UO22+) within 24 h [35].Due to the formation of nephrotoxic uranyl complexes such as carbonates and citrates, the primary damage induced by soluble uranyl ions is considered to be renal injury [36,37].
At physiological pH (7.4), Np(IV) and Np(V) may present simultaneously, and their biological behaviors are significantly different.In comparison to Np(IV) that forms complexes with Tf, Np(V)mainly exists as a free cation in the blood probably due to its low effective charge.One hour after the intravenous (iv) injection, more than 99% of Np(V) in the form of NpO2+is rapidly eliminated from the plasma, while approximately 20% Np(IV) still remains.The higher bioavailability of NpO2+through the kidney barrier contributes to the higher proportion of Np(V) (20%–40%) being excreted by the kidneys than that of Np(IV) (15%)[32,38,39].
For transplutonium elements, such as Am, Cm, the most stable valence is IIIin vivo[33].Similar as Pu, Am is concentrated in the plasma fraction, and nearly all are bound to transferrin.However,comparing to An(IV), the binding affinity of transferrin to Am(III)is weaker, which results in a faster clearance rate of Am(III) from blood [40,41].There are only less than 10% and 1% of Am injected stay in the blood 1 h and 24 h later, respectively.A few days after the parental injection, typically 80% or more Am accumulates in the liver (~50%) and bone (~30%) [40–42].
3.Actinide decorporation agents
For an applicable actinide decorporation agent, high binding affinity and selectivity, low toxicity, and good cell membrane permeability is required [43].From the coordination chemistry perspective, it is crucial to design a ligand with strong metal binding units and matched geometry of certain actinide ions to improve the actinide removal ability and also avoid the deficiency of important metal ionsin vivo, such as Zn2+, Co2+, Cu2+and Mn2+[7,44,45].In the past few years, various decorporation agents, including organic molecules and functionalized nanoparticles, have been designed, synthesized, and evaluated for their actinide decorporation performance [7,15,46-50].Thus, in the following section,the discussion will be divided into two parts, namely the molecular ligands and nanoparticles for actinide decorporation.
3.1.Ligands for actinide decorporation
The current inventory of molecular decorporation agents can be broadly classified into the following categories according to the selection of functional groups:
(1) Polycarboxylates.Ethylene diamine tetraacetic acid (EDTA) and diethylene triamine pentaacetic acid (DTPA) are valuable chelators for actinides decorporation.The DTPA salts are the only actinide chelators approved to use clinically, which could effectively remove Pu(IV) and Am(III)in vivobut show limitations in removing Np(V) and U(VI).
(2) Siderophores.The family of siderophores is applied to decorporate actinidesin vivo.Three types of binding subunits of catecholates (CAMs), hydroxamates, and hydroxypyridinonates (HOPOs), which are found in siderophores, are utilized to construct multi-dentate ligands for actinide decorporation.Among these candidates, HOPO ligands, such as 5LIO-1-Cm-3,2-HOPO, 5-LIO-(Me-3,2-HOPO), and 3,4,3-LI-(1,2-HOPO), show remarkable preponderance over catecholates (CAMs) and hydroxamates in effi-ciency and toxicity.
(3) Polyphosphonates.1-Hydroxyethane-1,10-diphosphonic acid(HEDP), as an authorized medicine for bone disease, is effective for uranium internal contamination.Other phosphonate ligands were also designed and synthesized, but their actinide decorporation performances were seldom investigated.
By using various types of backbones, such as linear, dendrimer,macrocyclic, or even calixarenes, a large number of ligands containing those functional groups were obtained.Among those, the well-established ligands and the latest reported ligands are mainly included in the following section Fig.2, where the solution chemistry, solid state coordination chemistry, and the decorporation efficacy of the selected ligands with different actinide ions are discussed in detail.Some previous reviews have done excellent jobs in summarizing the work in this field and the readers are referred to those references as well [7,51-57].
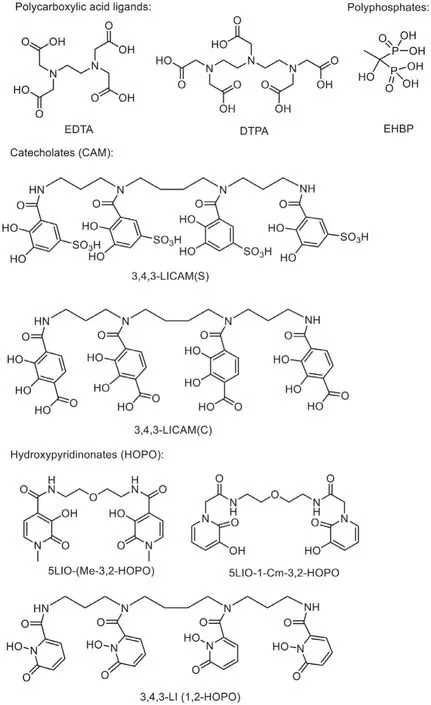
Fig.2.Structures of representative decorporation ligands.
3.1.1.Plutonium and thorium decorporation
Two representative polycarboxylate ligands are EDTA and DTPA.EDTA was first used as an analytical agent for calcium due to its high affinity for Ca(II) (logKCaL=10.28) in 1947 [58], and was then introduced in the antidotal therapy of90Y and144Ce [59].For actinides, EDTA is able to remove239Pu and241Amin vivo, but barely mobilizes uranium in kidneys and bones [60].This is owing to the higher complexation ability of EDTA toward Pu(IV) comparing to that of divalent cations such as Zn2+, Co2+, Cu2+and Ni2+(Table S1 in Supporting information) [61,62].However, the chelating ability of EDTA for actinides is apparently not high enough to outcompete the biomacromolecules such as transferrin that binds firmly with actinides (Table S1) [63,64].Consequently, H5-DTPA with five carboxylic acid groups was reported in 1954 [65].The stability constant of DTPA and Pu(IV) (logK=29.5) is increased by 3 orders of magnitude than that of EDTA (logK=26.4), resulting in improved decorporation efficiency and successful mitigation of renal damage.Hence, DTPA salts are authorized for actinide decorporation with a clinically acceptable dosage of 30 μmol/kg [65].Yet, their clinical administration is still limited due to low oral activity and high toxicity level caused by repeated injections [66].M.Jayet al.demonstrated the better actinide decorporation performance of DTPA-esters than pristine DTPA salts [67,68].It has also been reported that by encapsulating DTPA in novel drug delivery systems such as liposomes to modify the biodistribution of DTPA, improved plutonium removal efficiencyin vivocould be achieved [69–72].
A series of siderophores were used for actinide decorporation since the surface charge to ionic radius ratio of Pu(IV) is similar to that of Fe(III).Among the siderophore family, two types of binding moieties, catecholates (CAMs) and hydroxypyridinonates (HOPOs),exhibited high actinide binding affinity.Crystal structures of Th(IV)or Ce(IV) with bidentate catechol show that one metal center is bound to four ligands in the chelation mode (Fig.3) [7,73].The formation constant logβ110of catechol and Th(IV) is 17.72, proving the strong and selective binding of the ligand towards An(IV) [70].A large number of multidentate CAM ligands were designed and synthesized to decorporate actinides.Chenet al.introduced a series of CAM ligands, such as CBMIDA, 8102, 811, and FZ-82-4, that could remove thorium and plutonium from the targeted tissues [74–77].A vast majority of CAM ligands were reported by Raymondet al.Among these CAMs ligands, the formation constant of octadentate 3,4,3-LI(CAM) with Th(IV) is logβ110=47.71(8) (Table 1), which is clearly higher than those of DTPA and EDTA [78].In addition, those CAM ligands display a larger discrepancy in stability constants between An(IV) and M(II) (M(II)=Zn(II), Mg(II), Ni(II), Cu(II), Ca(II),and Co(II)) than DTPA, indicating its relatively higher selectivity of An(IV)in vivo(Table S2 in Supporting information) [79].
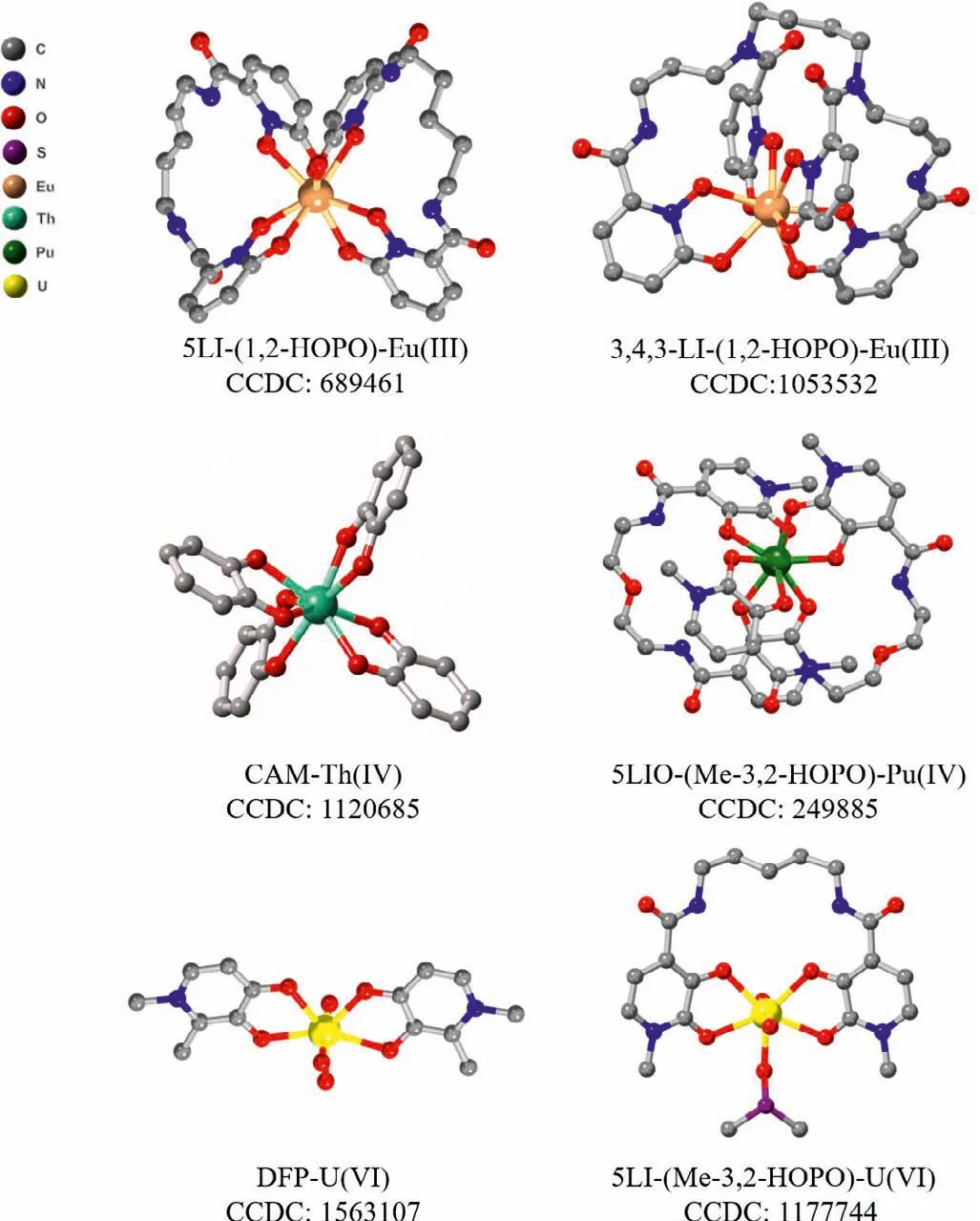
Fig.3.View of coordination modes between Eu(III), Th(IV), Pu(IV), and U(VI) with ligands of different denticity.Redraw from reported crystal data.
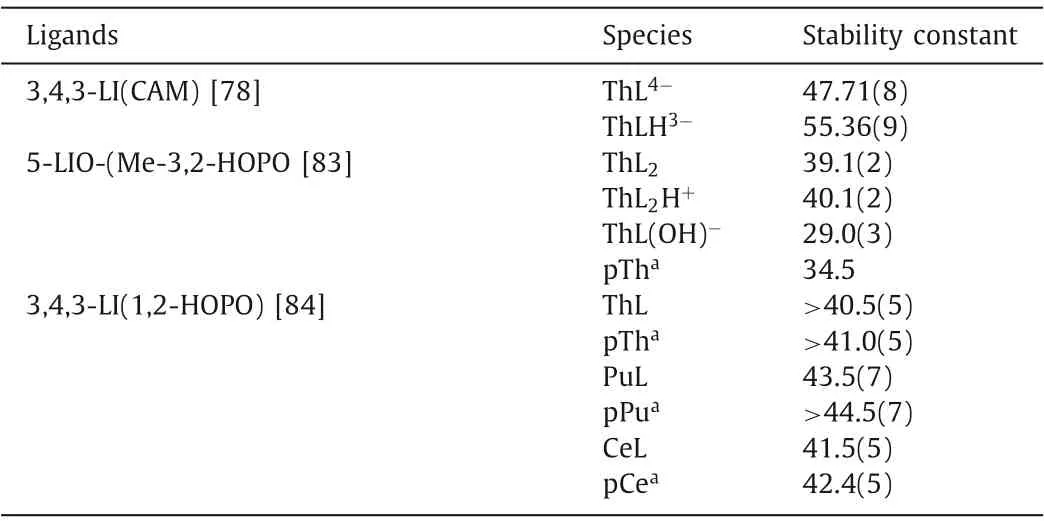
Table 1 The stability constants of CAM and HOPO ligands and actinide/lanthanide ions complexes.
Among siderophore ligands, hydroxypyridinonate (HOPO) ligand, generally adopts smaller protonation constants than bidentate catecholate and hydroxamate ligands, and therefore a higher proportion of the HOPO ligand is deprotonated at physiological pH (7.4), providing stronger classical Coulomb interaction with actinide ionsin vivo.Raymondet al.proposed that,viaamidation,the protonation constants of bidentate ligands could be further decreased by the introduction of intramolecular hydrogen bond between nitrogen atom from the amide unit and oxygen atom fromthe hydroxyl unit [80].Accordingly, three bidentate ligands, PR-1,2-HOPO, PR-Me-3,2-HOPO and PR-3,4-HOPO-N ligands were reported, where the intramolecular hydrogen bond is stronger in the former two than the latter.The protonation constants of PR-1,2-HOPO (4.96) and PR-Me-3,2-HOPO (6.12) are much lower than that of PR-3,4-HOPO-N (9.47) [81].The formation constants logβ140of three ligands with Th(IV) are 36.0(3), 38.3(3), and 41.8(5) for PR-1,2-HOPO, PR-Me-3,2-HOPO, and PR-3,4-HOPO-N, respectively (Table S3 in Supporting information).Similar as bidentate CAM ligands, in the structure of Ce-1,2-HOPO compound, the metal ion bonds to eight oxygen atoms from four bidentate HOPO ligands,forming a dodecahedron geometry [82].
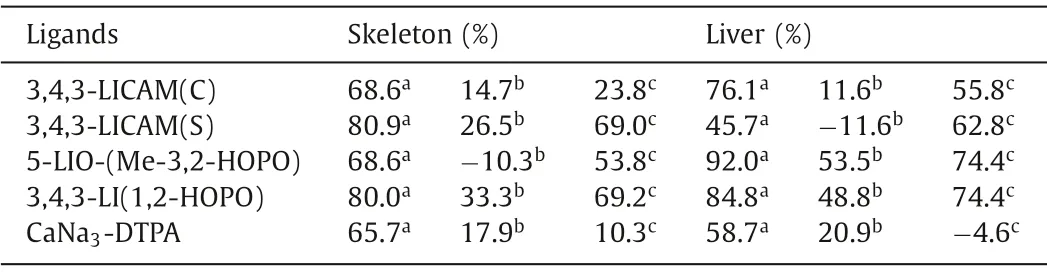
Table 2 The average Pu(IV) decorporation efficiency of DTPA, CAM and HOPO ligands.
Subsequently, Me-3,2-HOPO or 1,2-HOPO were mainly used as the chelation unit to obtain ligands with higher denticityviaamidation with various linkers.The pKavalues of those ligands vary from 4.9 to 7.02 [7].Among those ligands, 5-LIO-(Me-3,2-HOPO)and 3,4,3-LI-1,2-HOPO were screened for Pu(IV) decorporation.Considering the fissionable nature and biohazard of Pu(IV), Th(IV)and Ce(IV) are chosen as less or non-radioactive analogues for Pu(IV) due to their similar oxidation states, ionic radii, and coordination numbers [33].The tetradentate HOPO ligands tend to form 1:2 complexes with An(IV).The formation constants of 5-LIO-(Me-3,2-HOPO) were calculated as logβ120=39.1(2), logβ121=40.1(2),and logβ12–1=29.0(3) with Th(IV) and logβ120= 41.5(5) with Ce(IV) (Table 1), respectively [83].The octadentate HOPO ligands preferably form 1:1 complexes with An(IV).The formation constant logβ110of 3,4,3-LI-1,2-HOPO is>40.5(5) with Th(IV) and 43.5(7)with Pu(IV), respectively [84].The ionic radius of Ce(IV) and Pu(IV)is nearly identical and is smaller than that of Th(IV), therefore, the higher formation constants of Pu(IV)/Ce(IV) than Th(IV) for both ligands could be majorly ascribed to the higher surface charge of Pu(IV)/Ce(IV) (Table 1).For Th(IV), the pTh value of the octadentate ligand is 6 orders of magnitude higher than that of tetradentate ligand at the given condition despite of the similarity in their formation constants with Th(IV) [84].Crystal structure analysis reveals that both Ce(IV) and Pu(IV) metal center can be chelated by eight oxygen atoms from two tetradentate HOPO ligands (Fig.3)[85,86].
The Pu(IV) decorporation performance of CAM and HOPO ligands is presented in Table 2.For the mice that were intraperitoneally administrated with 30 μmol/kg ligand 1 h after being injected with238Pu(IV) intravenously, approximately 58.7% and 65.7%238Pu(IV) in the liver and skeleton was removed in the CaNa3-DTPA treated group [15].In comparison, 3,4,3-LI-CAM(S) and 3,4,3-LI-CAM(C) could significantly reduce 45.7% and 76.1% of238Pu(IV)in the liver, and 80.9% and 68.6% of238Pu(IV) in the bone, respectively.This result indicates that the hydrophilic 3,4,3-LI-CAM(S) has an advantage in reducing skeletal Pu, while the lipophilic 3,4,3-LICAM(C) is superior in the removal of Pu(IV) in the liver [15].For HOPO ligands, the group treated with the tetradentate 5-LIO-Me-3,2-HOPO yielded 92.0% and 68.6% removal efficiency for Pu(IV)in the liver and skeleton, respectively, while the octadentate 3,4,3-LI-1,2-HOPO achieved an even higher Pu(IV) removal efficiency of 84.8% and 80.0% in the liver and skeleton, respectively, indicating that octadentate ligand is more effective in decorporating An(IV)[15].Prolonging the ligand treatment time to 48 h resulted in reduced removal efficiency for most ligands [87–89].
In the oral administration experiments, both CAM and HOPO ligands are orally active while CaNa3-DTPA could not sufficiently remove238Pu in tissues possibly due to its high hydrophilicity[90,91].Specifically, 3,4,3-LI-CAM(S) and 3,4,3-LI-CAM(C) reduced 62.8% and 55.8% of238Pu(IV) from the liver, and 69.0% and 23.8%of238Pu(IV) from the bone, respectively [88].Moreover, both HOPO ligands can reduce 74.4% of238Pu(IV) from the liver and can remove 53.8% and 69.2% of238Pu(IV) from the skeleton by 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI-1,2-HOPO, respectively [90,91].
In wound contamination, 30 min after the intramuscular (im)injection of238Pu(IV), local im injection of 3 and 30 μmol/kg 3,4,3-LI-1,2-HOPO reduced 97% and 99% of Pu(IV) in the wounded site,respectively, comparing to the untreated mice.The ligand was still effective when extending the ligand administration time to 24 h(removal percentage in the wounded site was reduced by 76%)[23,92].Similar to Pu(IV), 86.1% of228Th(IV) was removed in the wound site in prompt administration, and 57% of228Th(IV) was removed by the im injected ligand 24 h after the initial im injection of228Th(IV) [27].
3.1.2.Uranium decorporation
Uranium could rapidly deposit in the targeted organs in a stable form of hexavalent uranyl ion [UO22+, U(VI)], further inducing irreversible renal damages and increasing risk of osteosarcoma[93–95].The U(VI) ion binds to 4 to 6 donor atoms in the equatorial plane perpendicular to the uranyl ion.From the perspective of coordination chemistry, EDTA and DTPA ligands could satisfy the isotropic coordination requirements of An(III) and An(IV), but may not fully occupy the coordination sites of UO22+on the equatorial plane [96].As shown in Table S1, the complex abilities of EDTA and DTPA toward U(VI) are lower than that of essential divalent metal ions, displaying poor selectivity in chelation toward uranyl ionsin vivo[61,97].Therefore, their decorporation performance of U(VI) is less than satisfactory.
Compared with DTPA and EDTA, tetradentate, hexadentate, and octadentate CAM ligands could coordinate with uranyl ion in the equatorial plane.The crystal structure of uranyl and tetradentate ligand 5LIO-(MeTAM) reveals that the four oxygen atoms from the ligand dominate four sites in the plane and the fifth site is occupied by solvent molecules, forming a typical pentagonal bipyramidal geometry [98].The equilibrium constants of tetradentate CAM ligands and uranyl were determined through spectrophotometric titrations, as shown in Table S3, and are clearly much higher than those of EDTA and DTPA [99,100].More recently, Penget al.rationally designed and synthesized a series of tetradentate and hexadentate CAM ligands with high affinity for uranyl(logβ110=29.96(3)~30.21(2)) using a flexibleβ-dicarbonyl as the linker [100].
The uranium decorporation efficacy was comprehensively investigated with multidentate CAM ligands, including tetradentate (2LI-, 4LI-, 5LI-) or BPCBG (N,N′-1,2-ethanediylbis[N-[(2,3-dihydroxyphenyl)methyl]]-glycine), hexadentate (3,4-LI-, Me-), and octadentate (3,4,3-LI-) ligands [35,101-108].Among these ligands,octadentate LICAM(S) displayed an obvious advantage in removing uranium deposited in the kidneys and bone by 61.1% and 66.9%, respectively (Table 4) [35].By increasing the molar ratio of the linear tetradentate ligand and uranium from 75 to 91, a higher removal efficiency was achieved [108].However, limited efficiency was observed when extending the treatment time to 48 h.Then oral activities of linear tetradentate ligands 4-LICAM(C), 5-LICAM(S), and 5-LICAM(C) were tested with different doses rangingfrom 30 μmol/kg to 200 μmol/kg.Results show that those ligands could hardly remove uranium from the bone, while no more than 50% uranium could be removed from the kidneys.Only ~18.8% of uranium was removed from the kidneys for the mice treated with 30 μmol/kg of 3,4,3-LI-CAM(S) [35,108].In addition, Chenet al.evaluated the performance of BPCBG.The cytotoxicity data proved that BPCBG exhibited lower cytotoxicity than DTPA-CaNa3.In vivouranium removal assays show that BPCBG could effectively decreased the uranium deposition in kidneys by 59%-69% induced by local im injection [101,102].In preclinical studies, CAM ligands were found to be nephrotoxic, and both 3-LI- and 3,4-LI-CAM caused severe damage to the kidneys, liver, and/or spleen, so further investigation of CAM ligands is restrained [7].
The multidentate hydroxypyridone ligands are promising decorporation ligands for uranyl.The solution thermodynamic study of bidentate HOPO ligands with uranyl show that they tend to form 1:2 metal:ligand complexes.The formation constant logβ120is 17.91 for PR-Me-3,2-HOPO and 16.46 for bidentate 3,4-HOPO(DFP) [109,110].The stoichiometric ratio in the crystal structure of the uranyl-DFP compound is consistent with the result of the solution thermodynamic study (Fig.3).For tetradentate HOPO ligands,a series of ligands with different linkers were evaluated for their chelating ability toward uranyl using the titration method.These ligands formed stable complexes with a metal to ligand ratio of 1:1 with different protonated states in aqueous solution.The main species found at pH 7.4 is [UO2L], and their stability constants of logβ110ranged from 12.5 to 14.9, which means that the variation of linkers does not significantly affect the metal binding affinity[109,111].Among those tetradentate ligands, 5-LIO-(Me-3,2-HOPO)with the linker of (-CH2CH2OCH2CH2-) gives the highest formation constant of logβ110=14.9(2) (Table 3) [83].Structural characterization of the coordination mode of uranyl complexes with tetradentate ligands revealed the typical planar coordination geometry of the uranyl unit (Fig.3) [111–113].
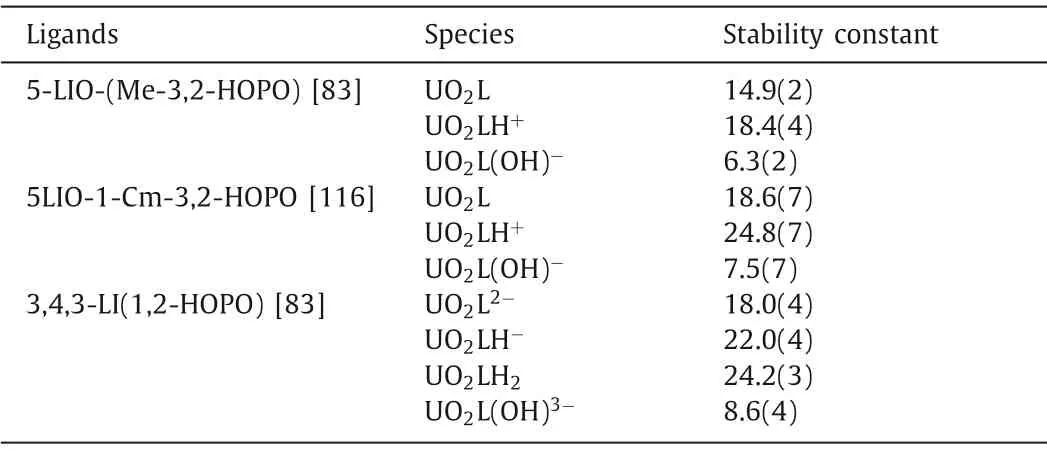
Table 3 The stability constants of CAM and HOPO ligands and uranyl complexes.
The complexation behavior of octadentate 3,4,3-LI-1,2-HOPO with U(VI) is similar to that of 5-LIO-(Me-3,2-HOPO), forming 1:1 metal:ligand complexes with a stability constant of logβ110=18.0(4) [80].The main species found at pH 7.4 is the deprotonated stoichiometric complex [UO2(L)]2-.The planar hexadentate ligand is predicted to be perfectly matching the coordination environment of uranyl, however, the decorporation efficacy of planar hexadentate ligand is not reported yet [114].Raymondet al.has reported that the toxicity of these tetradentate hydroxypyridinone ligands vary with the alteration of the linker and the toxicity increases in the trend of C4>C3>C6>C5.Among those linkers, 5LIO scaffold (-CH2CH2OCH2CH2-, 5LIO-) was determined to be the least toxic one [115].Eventually, two ligands of 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI(1,2-HOPO) were screened out as the optimal chelators for uranyl.5-LIO-(Me-3,2-HOPO) and 3,4,3-LI(1,2-HOPO)yielded 84.4% and 81.1% uranium removal efficiency in the kidneys with the intraperitoneal (ip) injection of ligand 3 min after the iv injection of U(VI), as illustrated in Table 4.However, prolongedtreatment of 48 h resulted in a decrease of efficiency to 38.8%for 5-LIO-(Me-3,2-HOPO).Both ligands were also orally active and 43.5% of uranium was eliminated from the kidneys in the mice by oral treatment with 100 μmol/kg 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI(1,2-HOPO) 3 min after iv injection of U(VI) [35,108].The only drawback of the two ligands is their limited uranyl decorporation in bones.This is partially because of the presence of intramolecular hydrogen bonds within those two ligands that have been designed to improve the ligand rigidity and promote the deprotonation of the ligand under physiological conditions so that stronger Coulomb interaction can be achieved between negatively charged ligand and positively charged uranyl ions, similar as discussed in the Pu decorporation section.However, strong intramolecular hydrogen bonds would weaken the binding affinity with uranyl from a thermodynamic perspective, which results in reduced formation constants values.To overcome this obstacle, Wanget al.introduced a new uranium chelator (5LIO-1-Cm-3,2-HOPO), in which the strength of intramolecular hydrogen bonds are reduced due to the presence of an additional methyl group between the amide and HOPO groups [116].The stability constants of the uranyl and 5LIO-1-Cm-3,2-HOPO complexes were determined to be logβ111=24.8(7),logβ110=18.6(7), and logβ11–1=7.5(7), much higher than those of 5-LIO-(Me-3,2-HOPO) (Table 4).DFT calculation results further elucidate the lower binding energy of UO2–5LIO-1-Cm-3,2-HOPO(-9.23/-9.21 eV) in comparison to that of UO2–5-LIO-(Me-3,2-HOPO) (-8.81 eV).Comprehensive cytotoxicity studies show that the cytotoxicity of 5LIO-1-Cm-3,2-HOPO is at the same level as ZnNa3-DTPA, but much lower than that of 5-LIO-(Me-3,2-HOPO).Moreover, 5LIO-1-Cm-3,2-HOPO achieved high U(VI) removal effi-ciencies of 86.8% and 47.9% in the kidneys and femurs, respectively,which is six times higher than the removal efficiency in the femurs of 5-LIO-(Me-3,2-HOPO) (8.0%).Similarly, in oral administration, 5LIO-1-Cm-3,2-HOPO exhibited a much higher U(VI) removal ratio in femurs of 30.5% than 5-LIO-(Me-3,2-HOPO) (3.5%), while their U(VI) removal percentages in kidneys were similar.
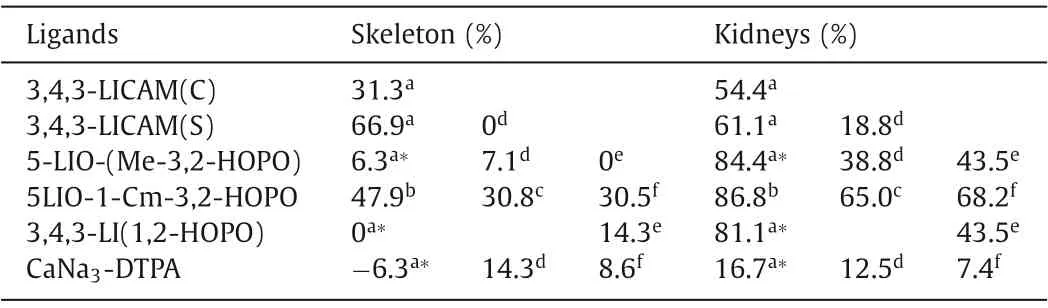
Table 4 The average U(VI) decorporation efficiency of DTPA, CAM and HOPO ligands.
Uranyl could form highly stable and insoluble complexes with hydroxyapatite (HAP) in bone, which is difficult to be excreted.A series of cyclic or linear polyphosphates, such as EHBP (ethane-1-hydroxy-1,1-bisphosphonate), DDMT (1,2-phenylene-dicarboxydiaminomethanetetraphosphonate), CDTT(1,2-diamino-cyclohexyl-tetramethylenetetraphosphonate), and BCPBP (benzo-cyclopentenebisphosphonate), were obtained and their uranium removal efficiency were evaluated [117–122].Lecouveyet al.has developed a simple and one-pot synthetic protocol for polyphosphonate ligands [118].Among these ligands, EHBP, as the optimal one, could reduce 76% of uranium from the kidneys[122–124].However, the nephrotoxicity of these ligands seriously limits their application [124].Taran and Ansoborloet al.screened out 23 dipodal and tripodal polyphosphates ligands among 96 candidates by the assistance of a chromophoric complex displacement procedure.Despite the high affinity of those ligands towards the uranyl ion, only limited decorporation efficiency was observed,and the dipodal bisphosphonate (3C) could prompt the uranium excretion by 55% from kidneys [119,120].More recently, Yeet al.designed and synthesized two tetraphosphonated pyridine ligands and demonstrated their high binding affinity of uranyl, however,the toxicity andin vivodecorporation study was not performed[125].
As early as the 1990s, Montagneet al.synthesized calixarene nanoemulsions and demonstrated their effectiveness in uranium decorporation, but some of these materials are hepatotoxic.Recently, Lecouveyet al.designed upper-rim polyphosphorylated calix[4]arenes for uranium decorporation, which shows similar uranyl chelating ability to the phosphonate ligand tris (HEDP),yet the uranium removal effectin vivohas not been evaluated[126,127].Phosphorylated pentapeptide was synthesized for uranium decorporation by Liet al.for the first time, which displayed much higher affinity for uranyl in comparison to nonphosphorylated polypeptides, and markedly improved the cell viability of HK-2 cells treated with uranium [128].
Besides the common chelating moieties such as catechol, hydroxamate, and hydroxypyridione, new bidentate ligands have been explored recently for the purpose of actinide decorporation.3-Hydroxy-2-pyrrolidinone (HPD), as anN-substituted pyrrolidinone, has recently attracted much attention in the pharmaceutical field because of its low toxicity.Wanget al.comprehensively studied its chelating ability and coordination mode with uranyl,as well as its removal efficacy of uraniumin vivo.Uranyl binds with two HPD ligands in solution with the formation constant of logβ120=20.7.In vivouranium decorporation assays show that 52%of uranium is removed from the kidneys by HPD, which is at the same level as DFP, while HPD exhibits a much lower level of cytotoxicity than DTPA-ZnNa3and DFP [129].2-Hydroxy-6-(propan-2-yl)cyclohepta-2,4,6-trien-1-one (Hino) was recently demonstrated to be an excellent candidate [130].Hino prefers to bind with uranyl in a stoichiometry of 3:1 and is able to selectively extract uranyl in the presence of essential metal ions.Furthermore, it could accelerate the excretion of uranium from the kidneys and femurs by the efficiency of 67.0% and 32.3%, respectively, which is much higher than that of DFP and HPD.These studies indicate that various bidentate ligands with high metal binding affinity could be utilized as new functional groups to construct advanced actinide decorporation agents.
3.1.3.Neptunium decorporation
Similar to U(VI), the stability constants of EDTA and DTPA for Np(V) are much lower than those of the essential divalent metal ions (Zn, Co, Cu, and Ni) (Table S1), and the geometry of EDTA and DTPA does not satisfy the coordination requirements of NpO2+[61,131,132].Therefore, their efficiency in removing Np(V)in vivois limited (Table S4 in Supporting information).CAM ligands also showed limited efficiency in eliminating Np(V)in vivo[7,15].There are extremely limited studies of the chelating ability and coordination mode of chelators with Np(V), probably due to its redox active nature which renders the related study difficult.Thein vivodecorporation efficiency of nine multidentate HOPO ligands towards Np(V) was reported [7].Among those, 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI-1,2-HOPO were the optimal ligands for Pu(IV)and U(VI), and their decorporation performance of Np(V) is discussed in detail here.As shown in Table S4, in prompt ip injection group, 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI-1,2-HOPO removed 88.8% and 40.6% of Np(V) from the liver and 31.6% and 42.1% ofNp(V) from the skeleton, respectively.In prompt oral administration group, 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI-1,2-HOPO reduced 52.4% and 19.0% Np(V) from the liver, respectively, but was nearly invalid in the skeleton [15].Though the reduction ratio of Np(V)from the liver was not high as that of Pu(IV), the bone decorporation performance of Np(V) is much better than U(VI).
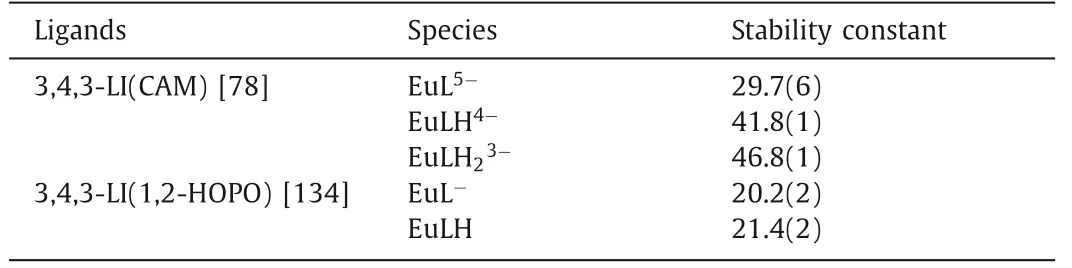
Table 5 The stability constants of CAM and HOPO ligands and Eu(III).
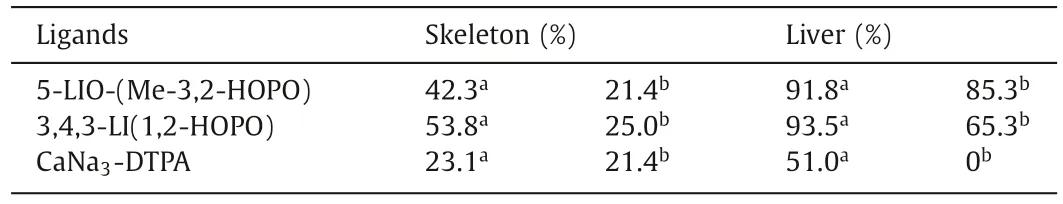
Table 6 The average Am(III) decorporation efficiency of DTPA and HOPO ligands.
3.1.4.Americium and transplutonium decorporation
Am isotopes are mostly products of neutron capture reactions,which adopt high chemo- and radiotoxicity, so Am(III) decorporation is also urgent.However, studies on the solution thermodynamics and coordination mode of chelators with Am(III) are limited due to its high radioactivity and difficulty in synthesis.To illustrate the solution thermodynamic behavior of Am(III), lanthanides ions especially Eu(III) was used as a surrogate for Am(III).As listed in Table 5, the formation of a 1:1 molar ratio complex of Eu(III) and 3,4,3-LI-CAM was observed in the spectrophotometric titration experiment and the formation constant of logβ110was calculated to be 29.7(6) [78].In contrast, the logβ110of octadentate ligand 3,4,3-LI-1,2-HOPO was 20.2(2), which is much smaller than the CAM analog.The tetradentate ligand 5LIO-1,2-HOPO forms 2:1 ligand:metal complex with a logβ120of 22.9, which is slightly higher than that of 3,4,3-LI-1,2-HOPO, but yields a lower pEu value at pH 7.4 [133,134].The single crystal structure of Eu(III)-5LIO-1,2-HOPO reveals the chelation of Eu(III) by two 5LIO-1,2-HOPO using eight oxygen donors [135].
For americium decorporation, ip injection of CaNa3-DTPA within minutes reduced 51.0% and 23.1% of241Am from liver and bone in mice, which is similar to the decorporation performance of238Pu at the same dosage, while no effective removal was observed in oral or 24 h delayed treatment (Table 6) [136].The ip injection of 3,4,3-LICAM(S) and 3,4,3-LICAM(C) 3 min after the iv injection of241Am resulted in a reduction ratio of only 34% and 56.8% in the liver, respectively [137].Compared with the CAM ligands, 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI-1,2-HOPO show remarkably improved decorporation efficiency of Am(III).The ip injection of 5-LIO-(Me-3,2-HOPO) and 3,4,3-LI-1,2-HOPO 1 h after the initial iv injection of241Am(III) reduced 91.8% and 93.5% of Am(III) in the liver and 42.3%and 53.8% of Am(III) in the skeleton, respectively [136].The oral administration of both ligands remove 85.3% and 65.3% of Am(III)in the liver and 21.4% and 25.0% in the skeleton, respectively (Table 6) [136].Besides, in skin decontamination, local im injection of 3,4,3-LI-1,2-HOPO could reduce the amount of Am(III) by 62% in the wounded site comparing to the untreated animals [138].
Moreover, the decorporation efficacy of transplutonium elements including Am, Cf, Bk, and Cm has recently been studied by Abergelet al., and their results indicate that the octadentate 3,4,3-LI-1,2-HOPO ligand could effectively remove those radionuclides [139].
It is noteworthy that the DTPA decorporation data for Th, U, Np,Pu, and Am discussed in this section are taken from those experiments where DTPA were used as the positive control group for direct comparison under identical conditions.There are many excellent researches that have investigated the decorporation efficacy of DTPA comprehensively [140–151].
3.2.Nano decorporation agents
Although molecular drugs have been widely investigated in actinide decorporation, the direct medication of drugs to patients may suffer from inherent deficiencies, including fast metabolism,frequent medicine intake, and low cell membrane permeability[45,152,153].Nano particles, particularly those with a moderate metabolic period, low biological toxicity, and high drug loading capacity, serve as promising drug candidates.In recent years,nanotherapeutic systems have represented a real breakthrough in medicine due to their low toxicity and chemical versatility[154,155].A large number of nanomaterials have been engineered and improved according to clinical requirements, certainly including chelation therapy.Different types of nanochelators, such as polymerides, polypeptide, deoxyribonucleic acid, and framework materials (MOF, COF,etc.), are involved in actinide decorporation,displaying high selectivity and biocompatibility (Fig.4) [47–50].
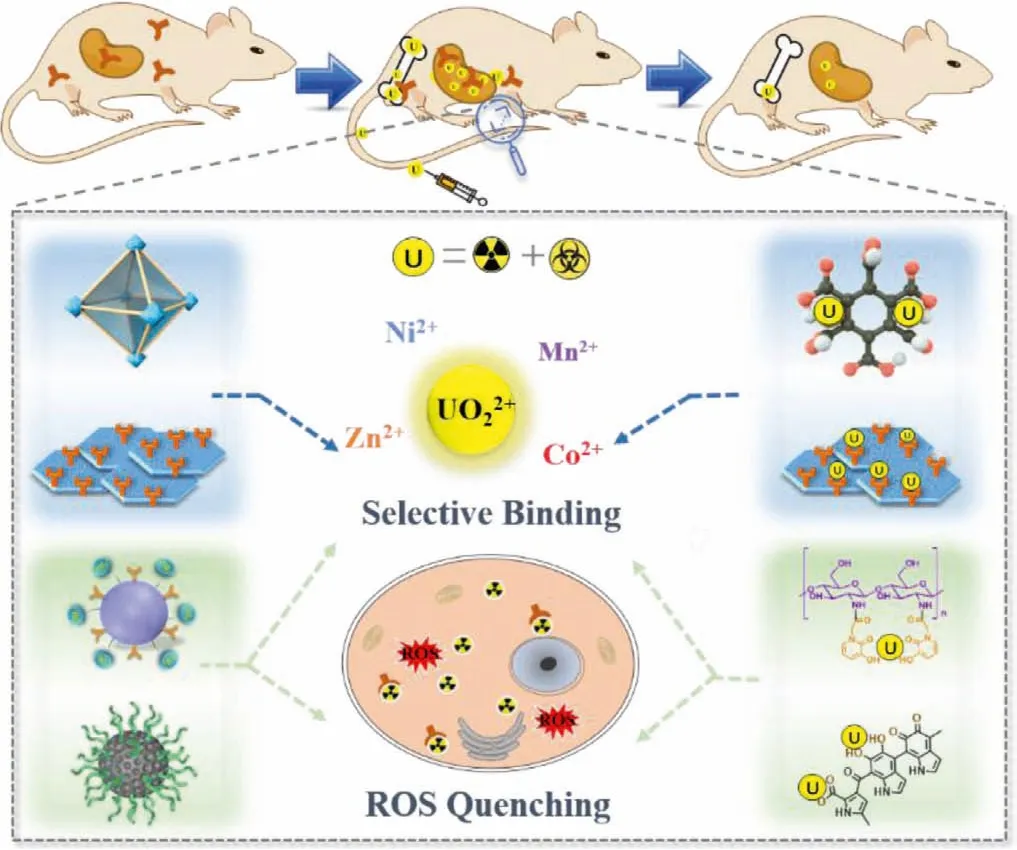
Fig.4.Illustration of metal-organic frameworks (UiO-66-(COOH)4-180 [48], covalent organic frameworks (CON-AO [49]), HOPO modified chitosan oligosaccharide(COS-HOPO [47]), and PEG grafted melanin nanoparticles (MNPs-PEG [50]) for actinides decorporation.
Due to the increasing interests and amounts of work published,there has been one recently reported review of nano radionuclide decorporation agents published inChin.J.Radiol.Med.Prot.[51].In this section, we will focus on the discussion of two types of nano materials, one is the exterior modified material obtainedviasurface fabrication and the other is the interior modified materials obtainedviapost-synthetic functionalization.
3.2.1.Exterior functionalized nano-chelators
Nanomaterials usually feature adjustable size and modifiable surface chemistry.The integration of nanotechnology and molecular biotechnology is attractive for researchers due to the enhancement of one specific function synergistically or the exhibition of multiple functions simultaneously.Current strategies include fabricating phosphonate, carboxylate, hydroxypyridinonates, or other metal binding units to various nanoparticles.
Early in 2006, Xuet al.reported a conjugate of dopamine and bisphosphonate (DA-BP), which was bound tightly to iron oxide to obtain a bisphosphonate modified magnetite Fe3O4nanoparticles.Combining the chelating effect of organophosphate and the inherent biocompatibility of iron oxide, a high level (99% and 69%) of uranium removal efficiency from water and blood was achieved [46].Di Giorgioet al.reported a methylphosphonated polyethyleneimine (PEI-MP) as candidate for uranium and thorium decorporation, which is able to load U and Th with a maximum capacity of 0.80 mg and 0.20 mg per mg of PEI-MP, respectively.The coordination mode was revealed to be similar to that of phosphonate with uranium or thorium by FT-IR and EXAFS [156].Subsequently, Zhanget al.reported a conjugate of DTPA and chitosan(WSC-DTPA)viaamidation, which exhibits better decorporation performance than DTPA-CaNa3[157].The additional renal protection effect of WSC-DTPA was also observed due to the ROS quenching ability of WSC.
Considering the above characteristics of chitosan nanoparticles and the high chelating ability of HOPO for actinides, Wanget al.developed hydroxypyridinone-grafted chitosan oligosaccharide nanoparticles (COS-HOPO, ~120 nm) as an agent with dual functions of uranium removal and ROS scavenging [47].Thein vitrometal ion adsorption experiments show that the material can selectively adsorb 92% uranyl from a solution containing excess competing divalent metal ions.The cytotoxicity assays show that COSHOPO is much less toxic than ZnNa3-DTPA and Me-3,2-HOPO to NRK-52E cells.The uranium removal efficiency in kidneys and femurs are 44% and 32%viaprophylactic administration, respectively.In addition, COS-HOPO is able to scavenge ROS induced by the uptake of uranium in NRK-52E cells, indicating that COS-HOPO could alleviate the oxidative stress induced by internal uranium expose.Hydroxypyridinone and catecholamide derivatized polyhydroxylated fullerene materials reported by Penget al.showed superior radical-scavenging capability and binding affinity for uranyl compared with the traditional chelating agent (DTPA) [158,159].Very recently, Diwuet al.proposed an ultra-small PEG grafted melanin nanoparticles (MNPs-PEG, 4–10 nm) as a bifunctional sequestering agent for uranium and thorium [50].Results confirmed that MNPs-PEG can selectively adsorb U(VI) and Th(IV) in the presence of excess Ni2+, Zn2+, Mn2+, Co2+, Mg2+and Ca2+at pH 4.0 and 7.4.Both in NRK-52E and AML-12 cells, MNPs-PEG displays acceptable cytotoxicity close to that of the DTPA-ZnNa3.This nanochelator could decorporate U(VI) by the level of 38.4% and 45.5%from kidneys and femurs, respectively, and Th(IV) by 42.8% from the liverin vivo.In addition, it can eliminate excess intracellular ROS and maintain the ROS at the normal level in cells exposed to U(VI) or Th(IV) by the combined effect of decorporation and ROS quenching, which was verified by the EPR andin vitrodecorporation section.
Those examples elucidate the advantages of nanocomposite in actinide decorporation with the ability of reducing the oxidative stress induced by internal exposure of actinide, which is beneficial to the treatment of contaminated individuals.
3.2.2.Interior functionalized nanochelators
Framework materials, such as metal-organic frameworks(MOFs) and covalent organic frameworks (COFs), are burgeoning classes possessing hybrid advantages of high surface area, highly ordered and constructable skeletons, and adjustable pore size,facilitating diverse platforms in catalysis, sensors, membrane separation and medicine.Their features of good biocompatibility provide possible usage for cancer therapy, biomedical imaging,and drug delivery [154].Horcajadaet al.proposed a pioneering oral detoxifying adsorbent agent based on MIL-127 for the efficient decontamination of salicylate [160].Moreover, functionalized MOFs and COFs are demonstrated to be highly efficient in capturing actinides in the environment, with high selectivity and capacity due to their steric arrangement of the chelating groups and optimized pore size [161,162].
Considering the excellent performance of nanoscale MOFs in the environmental decontamination and potential application as detoxicants, Diwuet al.prepared a carboxylic functionalized nanoscale MOF (UiO-66-(COOH)4–180, ~184 nm) forin vivouranium decorporation for the first time, where the carboxyl groups were preorganized in the channels, creating a coordination trap that enables the cooperative binding of uranyl [48].This material exhibited the fastest uranium uptake kinetics in solution by reaching the maximum adsorption ratio within 1 min, and strong ability of capturing uranyl from fetal bovine serum (FBS) by the level of 65% in only 5 min.UiO-66-(COOH)4–180 exhibited high selectivity for uranyl by absorbing 89.3% of uranyl in the presence of an excess of Ni2+, Zn2+, Mn2+, Co2+and Mg2+, whereas no more than 10% of those essential divalent ions were captured.The cytotoxicity of this material was determined to be at the same level as ZnNa3-DTPA.A series ofin vivoassays were performed, and the results indicate that this nano-MOF could accumulate in the targeted organ of kidney, and is capable of sequestrating uranium in blood and reducing uranyl deposition in kidneys and femurs up to 55.4%and 36.5%, respectively, showing superiority towards the polycarboxylate agent ZnNa3-DTPA.
Pursuing this strategy, another two-dimensional (2D) covalent organic nanosheet (CON) was rationally designed by Diwuet al.[49].In particular, this 2D material is functionalized with amidoxime (AO), a specific uranium binding ligand extensively used for the extraction of uranium from seawater.The attachment of AO groups to the open 2D structure makes them more accessible to capture uranium in solutions.More importantly, the AO groups are sterically arranged in order, which is beneficial for the selectively binding of uranyl according to the results of competitive adsorption assays.As a result, CON-AO could capture 85.4%-94.5% of uranyl ions and no more than 3.78% in the presence of divalent ions (Mg2+, Ca2+, Co2+, Mn2+, Ni2+, and Zn2+) from a mixture solution, in which the concentration of divalent ions was 5 folds that of uranyl ion.Furthermore, the cytotoxicity level of CON-AO to NRK-52E cells is similar as DTPA-ZnNa3.The results ofin vivoassays for both prophylactic and prompt administration show that CON-AO can reduce the amount of uranium in kidneys by 27% and 50%, respectively, which is higher than ZnNa3-DTPA under identical experimental condition.
Those work demonstrates that the preorganization of metal chelating groups in the interior space to match the coordination of certain metal ions will facilitate the selective binding ability,which is an obvious advantage forin vivodecorporation.Though the clinical usage of nano drugs is still facing many challenges,such as biosafety,in vivometabolism, and biodegradability, their advantages endow them as a research hot spot in the field actinide decorporation.
4.Conclusion
The research field of actinide decorporation represents an important and eye-catching hot point for the scientists and general public.The review presented herein contains the latest development of chelators for actinide decorporation and a brief overview of the optimal reported ligands in the past, with a general analysis of their decorporation efficacy, coordination feature, solution thermodynamics, adsorption behavior,etc.From those studies, it is concluded that the solution thermodynamic parameters of logβand pM values of a ligand are important to assess its selective binding ability to certain actinide ions, but does not guarantee excellent decorporation performance probably due to toxicity and pharmacokinetic reasons.It is important to note that tetradentate and octadentate ligands are effective for both An3+,4+and AnO2+,2+, despite their different biodistribution and metabolism patterns, and HOPO ligands seems to be the most promising ones currently, but there are still a lot of room for improvement.The nano decorporation agent represents a brand-new family of material, where the combination of many functional moieties together with various metal chelating groups arranged exteriorly or interiorly, exhibit clear advantage in organ targeting, low toxicity, and high binding selectivity,which are promising in overcoming the drawbacks of molecular agents.
Some future research directions that we think is promising in promoting the actinide decorporation performance are provided here.
Design of molecular ligands: The current inventory of decorporation ligands still suffer from drawbacks of high toxicity, fast metabolism, or limited removal ability.Therefore, new ligands with optimized binding group or geometry are highly desired.For the design the molecular ligands, the geometrical match of certain actinide ion and not with transition metal ions is critical for selective binding, which could be tailor-made based on the coordination stereochemistry, bond length, bonding nature,etc.The lipophilicity and the hydrophilicity are also important parameters for the pharmacokinetic optimization of the ligand.It is noteworthy that the formulation optimization of the current developed ligands is of equal importance as the design of new ligands, where the alteration of targeting organs and prolonged retention time are also beneficial to improve the decorporation performance.Besides,given the current understanding of the decorporation agents, it is possible to build a database and design the optimized ligand from the calculation results based on machine learning, which has already been utilized in the invention of new drugs with highly effi-cient outcomes [163–166].
Design of nano agents: Current research of nano decorporation agents is still at the initial stage.By the interior and exterior modifications, some ligands, such as carboxylate and phosphonates that have been restrained for actinide decorporation due to either high toxicity or poor efficacy, might be utilized again here with improved selectivity, reduced toxicity, and maybe better organ targeting efficiency.Given the vast selection of the nanoparticles (polymers: MNP, WSC, COS,etc.; porous framework: MOFs, PAFs, COFs,etc.) and functional groups, it is expected that a large variety of framework materials will be found with high radionuclide removal efficacy, low toxicity, and high oral activity.It is also possible to load various kinds of decorporation ligands, ROS quenching moieties, organ targeting units, or even fluorescence probes for multimode decorporation simultaneously.
Application of decorporation agents: The outcome in this field such as new actinide chelating ligands and nanoparticles could also be used in other radiochemistry fields in general andvice versa.For instance, the advantages of highly specific actinide binding ability endow those agents as good actinide chelating groups that may be useful in solvent extraction of actinides during the nuclear fuel recycling processes, in uranium ore mining and sea water extraction, and in the decontamination or depletion of actinide in the environment.What is more, those decorporation agents can serve as bifunctional chelator (BFC) or carriers/cargos for the development of novel radiopharmaceuticals using radioactive f elements such as Lu-177, Ac-225, Th-227, due to their low toxicity and strong f-element binding capability.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The work was supported by grants from the National Natural Science Foundation of China (Nos.21976127, U2167222, 21790370,22106116), the Natural Science Foundation of Jiangsu Province (No.BK20190044, BK20210736), a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and China Postdoctoral Science Foundation (No.2020M681716).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.04.017.
 Chinese Chemical Letters2022年7期
Chinese Chemical Letters2022年7期
- Chinese Chemical Letters的其它文章
- Professor Zhifang Chai: Scientific contributions and achievements
- Stable isotope labeling of nanomaterials for biosafety evaluation and drug development
- Emerging nanozymes for potentiating radiotherapy and radiation protection
- Recent progress of astatine-211 in endoradiotherapy: Great advances from fundamental properties to targeted radiopharmaceuticals
- Recent development in selective Tau tracers for PET imaging in the brain
- 64Cu radiolabeled nanomaterials for positron emission tomography(PET) imaging