
固固转化反应硫正极的研究进展
2022-07-07 03:32王久林
储能科学与技术 2022年6期
张 弘,张 阳,赵 耀,王久林,2
(1上海交通大学化学化工学院,上海200240;2新疆大学化学学院,新疆 乌鲁木齐830046)
随着时代和科技的快速发展,人类对煤、石油、天然气等传统能源的过度消耗导致在全球范围内出现了传统化石能源日益匮乏的能源危机以及温室效应和严重的环境污染等生态问题,因此发展新型清洁能源的要求日益迫切。近几十年来,作为新能源(太阳能、风能、潮汐能等)发展利用的重要一环,以可充电二次电池为代表的电化学储能因具有高效、高能量密度、清洁和受环境地域影响小等诸多优势而受到广泛关注,先进可充二次电池技术的研究一直是全球科研工作者不断追逐的热点[1-2]。其中锂离子电池(LIBs)的商业化开启了二次电池领域的新篇章,但锂离子电池由于其自身正负极理论容量的限制,已无法满足新兴市场不断增长的能量密度需求[3-4]。为了克服LIBs能量密度不足的限制,寻求其他具有高能量密度的电池体系至关重要。其中锂硫电池(Li-S)被认为是最具潜能的二次电池技术之一[5],作为正极的硫单质理论比容量高达1675 mAh/g,Li-S的理论能量密度为2600 Wh/kg,约高出常规LIBs 5 倍,此外自然界中稳定存在的硫单质还具有资源丰富、成本低廉、环境友好等优点[6-8]。
尽管如此,目前Li-S的商业化仍存在一些阻碍。对于硫正极来说,虽然,硫单质的理论储锂容量大,但是硫及其最终放电产物的电导率较低,其电子绝缘性严重制约了活性材料的利用[9];其次单质硫及其最终放电产物硫化锂(Li2S)的密度差别较大,分别为2.07 g/cm3和1.66 g/cm3,从而导致在电池充放电循环过程中会产生约80%的体积变化,正极结构被破坏造成不可逆的容量损失,影响循环稳定性[10]。对于活泼的锂金属负极来说,在循环过程中会不断消耗电解液,在界面处形成电子绝缘但离子导通的固体电解质界面(SEI)膜,降低电池的使用寿命;再者,液体电解液与锂金属接触时稳定性差,因锂金属粉化、膨胀和枝晶生长引起的热失控所带来的安全隐患仍然是非常严重的问题[11-12]。
此外,在循环过程中硫氧化还原反应产生的中间产物可溶多硫化物的穿梭效应是限制Li-S电化学性能和循环稳定性的关键问题之一。目前,Li-S中使用最广泛的电解液体系是醚类电解液,中间产物多硫化物在其中具有较高的溶解度。因此,硫与最终放电产物Li2S 之间的氧化还原转化经历了“固-液-固”的过程[10]。多硫化物在电解液中的溶解使得硫和Li2S 的氧化还原反应能够连续进行,一方面提高了硫的利用率,但另一方面溶解的多硫化物在反应过程中会向阳极侧迁移引发穿梭效应,绝缘产物Li2S会逐渐堆积在锂负极表面,导致了库仑效率低、电解液消耗、活性材料不可逆等损失。最终造成Li-S的实际能量密度有限,循环寿命短[13],不能满足实际应用需求。
为了解决多硫化物溶解穿梭问题,许多研究者提出了各种策略。一种是通过采用多孔的载体材料对硫进行物理吸附。这一概念早在2002 年就得以报道[14],2009年后引起了广泛关注[15]。将硫与活性炭或有序介孔碳复合,碳骨架结构有效地提高了绝缘硫的电化学活性,多孔结构的吸附效应抑制了多硫化物的扩散损失。这种方法可以提高正极的整体电子电导率,同时引入碳基体使比表面积增加,为氧化还原反应创造了丰富的反应位点,从而显著提高了电化学性能。但是上述碳材料对硫的限制是物理作用,由范德华力主导,在强电场和浓度梯度的作用下,这些作用力不足以将多硫化物限制在导电骨架中,因此进一步的策略是对载体进行化学修饰,以实现化学吸附。一些材料如有机聚合物等[16-17]在增强吸附力的同时,还可以促进离子在正极-电解质界面的转移,加速Li2S 的成核。此外还可以在硫基正极和隔膜之间构建阻挡层,或用各种涂层改性隔膜,阻碍多硫化物向负极扩散[10,18]。
虽然上述方法可以通过部分捕获多硫化物来抑制穿梭效应带来的问题,但还是存在反复的固-液相转化过程,不能完全消除可溶性长链多硫化物的扩散和迁移的问题。另外,常见的碳酸酯类电解液也会与溶解的多硫化物发生反应[19]。然而,“固-固”转化反应的硫正极可以从源头上解决多硫化物的产生[20]。固固转化反应[21]是指硫到多硫化物再到Li2S之间的固相转化,没有长链多硫化物的溶解过程。欲实现固固转化反应,需要在分子尺度上为绝缘硫活性物质构建电子/离子传递通道。主要有以下几种实现途径:一种方法是使用硫基材料,通过将硫限制在微孔结构的骨架内,阻止电解质渗透到正极;另一种方法是将硫与有机化合物共价结合,改变反应路径,从而阻止溶解过程[22-23];此外用固体电解质代替液体电解质,对应硫正极反应也是固固转化机理[24-27]。此外,还有一种“准固态”转化反应可以综合抑制穿梭效应和保证硫利用率[28]。在“准固态”转化反应中,多硫化物的溶解程度介于“固-液-固”和“固-固”转化之间,硫直接被还原为溶解度远低于长链多硫化物的短链多硫化物,再进一步转化为Li2S。“准固态”转化过程中,代表长链多硫化物生成的平台变短甚至完全消失。不同转化途径的典型充放电曲线见图1(a)~(c),传统的“固-液-固”沉淀-溶解转化反应具有两条平台曲线;“固-固”转化通常表现为单坡型;对于“准固态”转化,通常表现为在2.1 V 左右先有一个短的坡面,然后是一个平台[29]。“准固态”转化同时包含了另外两种反应路径的特征,为Li-S的进一步发展提供了一个新的研究方向。
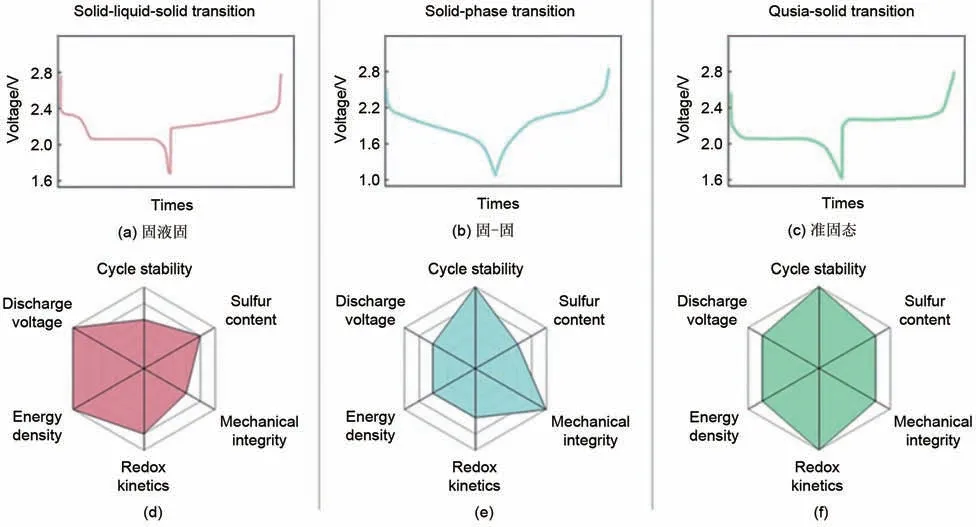
图1 Li-S电池不同转化路径的特征充放电曲线和常见性能指标总结[29]Fig.1 Summary of characteristic charge/discharge curves and common performance metrics for different transition pathways in Li-S batteries[29]
下文主要对硫正极的“固-固”转化反应机制进行综述分析,对比了不同策略的优缺点,总结了固固转化反应的硫正极与固态电解质的协同作用,并对其未来发展方向做出了展望。
1 固-固转化反应机制
“固-固”转化过程没有可溶性长链多硫化物的溶解,从而消除传统单质硫电极“固-液-固”反应过程中多硫化物溶解穿梭带来的不良影响。固固转化反应还具有以下特点:可使用商业成熟且相对廉价的碳酸酯类电解液;在容量-电压曲线上表现为单一平台;理论上电解液用量少、循环稳定性好、库仑效率高、自放电低甚至无自放电等。
1.1 微孔碳限硫
硫碳(S/C)复合材料最初是为了提高硫正极的导电性,以实现高硫利用率。在众多结构各异的碳基材料中,微孔碳(MC)孔径小于2 nm,可以在硫和电解质之间形成物理屏障,使硫进行固相转化反应。早在2002年,Wang等人[14]将单质硫填充在活性炭纳米孔和微孔中,在凝胶电解质中实现了固固转化反应,表现出单平台放电曲线。2010 年,Zhang等人[30]通过混合热处理,将单质硫以高度分散的状态填充在碳球狭窄的微孔中,如图2 所示,硫正极的电化学反应过程被限制在微孔内,研究发现在首次循环后,微孔碳内部形成了吸收能较低的特殊复合物,从而增强了复合正极的电化学稳定性,在400 mA/g电流密度下,硫含量为42%(质量分数,下同)的复合材料循环500 次后的可逆容量约为650 mAh/g。此后,硫/微孔碳(S/MC)复合材料因其高硫利用率和良好的可逆性而受到越来越多的关注。
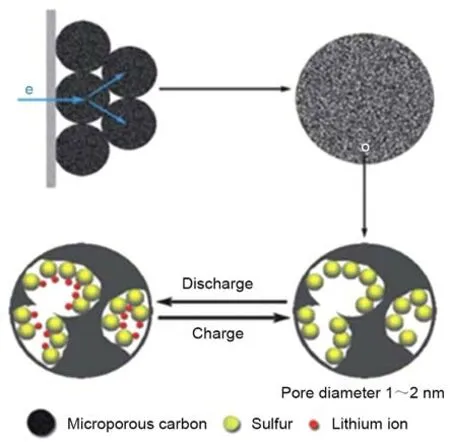
图2 硫碳复合正极微孔内约束电化学反应的示意图[30]Fig.2 Scheme of the constrained electrochemical reaction process inside the micropores of the sulfur-carbon composite cathode[30]
Xin 等人[31]发现,在孔径约为0.5 nm 的碳基体中,由于空间限制作用单质硫以S2~4小分子的形式存在,而不存在S8环。硫的转化反应在S2~4短链多硫化物与Li2S之间进行,解决了长链多硫化物溶解引起的穿梭问题。此后,Li等人[32]通过碳微孔内限制的S2~4小分子和分散在表面的自由S8分子的两种复合材料的对比,进一步阐明了碳的微观结构、硫的存在形式和电解质类型协同作用的重要性,被填充在微孔中的硫在醚类和碳酸酯类电解液中都能很好地工作,而分布在碳基体表面的硫只能在醚类电解质中工作。结论表明这些微孔通过尺寸限制抑制了电解液溶剂分子向硫正极的渗透,从而实现了硫正极的固固转化,同时抑制了多硫化物与碳酸酯分子之间的副反应。
但又有研究表明,对于一些碳孔径足够宽可以容纳S5~8等大分子硫的超微孔碳材料,与硫复合后仍可实现固固转化反应[33]。此外,另外的一些研究证实,环状S8在高温下很容易碎裂成气态的无环短链碎片,其链长随温度的升高而减小[34],所以550 ℃以下,S2~4小分子的含量极低[35]。在之前的报道中,大多数S/C复合材料的熔融渗透温度大约在300 ℃[36],所以可以合理地推测在硫的固固转化过程中,并不是一定需要形成小分子硫S2~4来代替长链多硫化物,关键是要有足够小的微孔避免硫材料与电解液溶剂分子的接触。
还有一种观点认为,S/C复合正极表面形成具有离子导电性的正极电解质界面(CEI)膜,是孔径大于2 nm的碳材料仍能引发固固转化反应的原因。Markrich 等人[33,37]进行了一系列的实验来验证这个观点,通过X 射线光电子能谱(XPS)表面分析证实了在初始放电过程中,独特的不可逆高容量代表了CEI膜的形成。此外根据氮气吸附等温线数据,当60%硫填充微孔时,氮气分子无法进入硫填充的微孔说明CEI膜阻止了电解质溶剂分子进入微孔,锂离子与硫的固态反应发生在无溶剂环境中。在初始循环期间,CEI膜的形成过程受电解液成分和放电截止电压的控制,与碳主体的多孔结构关系不大。
Xu等人[38]将不同比例的硫碳混合材料在600 ℃进行热处理,使单质硫气相分解成的S2小分子渗透进0.5 nm的碳微孔中,其中31%硫含量的S/C复合材料在4020个循环后仍然稳定保持了600 mAh/g的比容量。含硫碳酸盐成分的CEI膜会将S2小分子截留在碳基体的孔隙中,阻止其与电解液进一步反应。Wang 等[39]进一步研究了采用商业碳酸酯类电解液(1 mol/L LiPF6/EC/DMC)的S/MC 正极上CEI膜的形成机理和成分。结合模型化学反应和密度泛函理论(DFT)计算研究发现:由于Li+对EC 溶剂的优先溶剂化,乙烯基碳酸锂(LEMC)和甲基碳酸锂(LMC)是CEI 的主要有机成分,而Li+和EC 的相互作用有利于多硫化物与EC 分子的羰基碳结合。从而在正极-电解质界面形成如图4 所示的独特的有序电化学双层(EDL)结构。

图3 机理示意图:(a)“固--液-固”转化反应;(b)由于形成的正极CEI膜导致的“准固态”转化反应[33]Fig.3 Schematic diagram of mechanism:(a)“solid-liquid-solid”conversion reaction;(b)“quasi-solid-state”conversion reaction due to the formation of cathode CEI films[33]
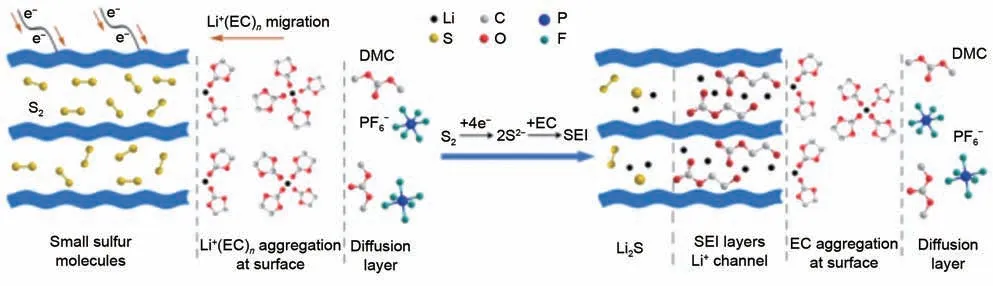
图4 Li-MC/S电池在初始放电过程和正极CEI膜形成过程中MC/S正极的EDL结构[39]Fig.4 EDL structure of MC/S cathode in Li-MC/S battery during initial discharge and the formation of CEI films[39]
总结来说,S/MC 复合正极实现固固转化反应抑制穿梭效应的合理解释观点是微孔碳通过小孔径限制硫分子大小、低孔隙率减少硫与电解液接触、形成CEI膜。但是通过微孔结构限制硫实现固固转化反应具有一些缺陷性。首先,碳基体孔隙率低会导致硫负载量受到限制;其次S/Li2S的电子导电性差,孔径尺寸的增加会影响硫利用率。所以孔径的选择上应该在高硫负载量和高硫利用率之间进行权衡。此外,到目前为止,在CEI膜形成机理的研究中采用的碳基体孔径具有微孔到大孔的广泛分布,电化学性能与孔径之间的关系仍不明确。需要系统地研究固固转化反应中的尺寸效应;同时,电解质和硫材料之间的反应以及原位形成的CEI膜的长期稳定性也需要进一步研究。
1.2 有机聚合物固硫
有机聚合物固硫的典型代表是硫化聚丙烯腈(SPAN 或S@pPAN)[23],硫分子与热裂解的聚丙烯腈(类聚乙炔结构pPAN)之间形成共价键或络合键,也可能存在硫对聚乙炔掺杂,致使S@pPAN 具有良好导电性;经过几次充放电循环后,二者之间的相互作用减弱甚至消失,导电性的pPAN为硫分子提供电子/离子快速传递通道,从而实现固固转化反应。自2002年首次报道S@pPAN以来,在不同的合成路线、不同的结构设计和电解质优化方面取得了广泛的进展。
早期有研究表明,聚丙烯腈(PAN)在热处理过程中会逐渐转化为具有吡啶-N 单元的环状结构[40]。与此同时,硫通过C—S键与π-共轭碳骨架共价结合[41]。为了阐明硫和PAN的复合形式以及循环过程中的结构演变,Fanous 等人[42]推导出了一个复杂的S@pPAN 结构,其中硫的主要存在形式为低聚硫化物、2-吡啶基硫代酸盐和硫酰胺。根据XPS和傅里叶红外光谱(FT-IR)图分析结果,得出硫只与碳成键的结论。Wei等人[43]提出硫以S3/S2小分子的形式存在,并共价连接到含有吡啶-N的碳主链上。在碳酸酯类电解液中多硫化物的穿梭从根本上受到抑制,从而可以达到优异的循环性能。循环伏安曲线进一步证明,反应涉及两电子转移过程,Li2S是唯一的放电产物,这解释了S@pPAN正极的高比容量。
研究者们一直致力于研究和优化S@pPAN 的反应机理和分子结构[44]。电解液组分优化和锂负极保护也有助于显著提高电池性能[45]。尤其是,碳酸酯类电解液在固固转化中起着重要作用[46]。Yang等人[47]在S@pPAN 正极体系中使用氟代碳酸乙烯酯(FEC)代替碳酸乙烯酯(EC)作为电解液,可以在正极上形成稳定的富含氟化锂(LiF)的CEI膜,控制甚至消除硫活性组分从S@pPAN 内部向表界面扩散或迁移,实现稳定固固转化反应,获得长寿命硫正极。在电流密度6 C 充放电下经过4000 次循环后比容量为1261.2 mAh/g(按硫计算),对应高容量保持率96.8%。
Wang等人[48]又对S@pPAN的超额初始比容量进行了深入研究。结合核磁共振(NMR)、电子顺磁共振(EPR)和理论模拟的结果,发现在第一次放电中,初始S@pPAN中的S—S键断裂,生成硫基自由基,可以与吡啶骨架上电子离域的硫基形成共轭结构(自由基S@pPAN),该共轭结构可以通过Li耦合电子转移过程与Li+反应,可逆地形成离子配位键(图5)。需要注意的是,在充电过程中,锂离子化S@pPAN 转变为共轭自由基S@pPAN,而不是初始S@pPAN 的结构,这也说明了初始放电曲线与以后循环的不同。Jin 等人[49]基于非原位固态核磁共振和XPS 的结果报道了一种新的共轭双键锂离子存储机制。他们将首次循环后的不可逆容量损失归因于S@pPAN聚合物骨架中的剩余锂。Li+除了与单质硫反应外,还会与PAN骨架中的C=N和C=C基团生成Li—C—N—Li 和Li—C—C—Li,产生多余容量。并且骨架中残余Li+也有利于提高整个正极的离子电导率和降低电极极化。Wang 等人[50]通过S@pPAN/CNT 电极证明了硫化聚合物骨架中存在残余锂,通过一系列的表征测试,发现硫以短链S2/S3的形式存在,通过可逆的C—S/S—S 键实现S 和Li2S 之间的转化(图6)。同时前线分子轨道(FMO)分析表明,S@pPAN 的锂化和硫原子从PAN骨架的脱离可以使聚合物主链更具导电性。

图5 SPAN储存锂的反应途径示意图。(a)初始SPAN;(b)具有共轭结构的自由基SPAN;(c)~(d)SPAN2-+2Li+;(e)SPAN3-+3Li+;(f)SPANn-+nLi+[48]Fig.5 Schematic illustration of the reaction pathway for SPAN to store lithium.(a)pristine SPAN;(b)radical SPAN;(c)-(d)SPAN2-+2Li+;(e)SPAN3-+3Li+;(f)SPANn-+nLi+[48]
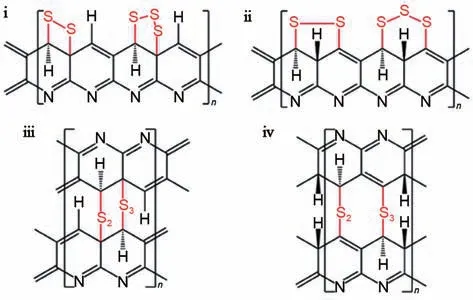
图6 SPAN的化学结构[50]Fig.6 Chemical structures of SPAN[50]
S@pPAN 正极在碳酸酯类和醚类电解质中均具有优异的循环稳定性,但很难有效提高S@pPAN中的硫含量,大部分情况下硫含量小于50%。有限的硫含量和相对较低的工作电压极大地削弱了以S@pPAN 为正极的Li-S 在能量密度方面的优势,能量密度与循环稳定性的不可兼得阻碍了其实际应用。为了克服这一局限性,开发高硫含量、高负载量、低E/S 比的S@pPAN 正极至关重要。同时,S@pPAN 的具体分子结构、反应机理和储锂机制仍存在争议。为了设计出更好的S@pPAN 复合材料电池体系,还需要对其结构和工作原理进行更进一步的基础研究。
除S@pPAN 外,其他类型的有机物固硫也可以实现固相转化。2013 年,Duan 等人[51]报道了一种碳炔多硫化物正极,小分子Sm(1≤m≤4)与导电碳骨架相连,在碳酸酯类电解液中显示出约1.8 V的单一放电平台。其中硫含量约为54.1%的正极材料在0.1 C电流密度循环200次后,可获得960 mAh/g的可逆容量。除了碳炔多硫化物,Du 等人[52]还报道了硫化石墨烯(SGDY)作为活性材料实现正极的固相转化(图7)。由于S8和石墨烯的高活性碳-碳三键之间的反应,硫以短链硫化物存在,共价键和物理约束使SGDY能够限制多硫化物,并与酯基电解质相容。因为共价有机多硫化物具有稳定的循环性能和可调硫含量的特点,为构建高性能正极材料指明了新的方向。

图7 简单的热合成工艺制备的SGDY的示意图[52]Fig.7 Schematic diagram of the preparation process of SGDY by using a simple thermal synthesis procedure[52]
尽管目前关于进行固固转化反应的硫化有机聚合物的报道较少,但基于有机聚合物材料的丰富性和结构的灵活性,未来可能会有更多关于此类复合材料的研究。
1.3 无机杂原子固硫
当硫与无机杂原子形成共价键的化学相互作用足够强时,结合合适的碳基体和电解质,可以实现硫的固固转化[53-55]。与硫相比,它的同系物硒(Se)和碲(Te)具有更高的电导率。由于Se/Te 原子的p轨道较高,在掺杂进硫正极形成复合材料后,可以在硫和Li2S的电子结构中引入更多的能态,从而有利于提高本征电导率,使反应动力学增强[56]。
2015 年,Li 等人[57]制备了一系列Se 掺杂的S1-xSex/C (x≤0.1)复合正极材料。研究证明Se 元素被均匀掺杂,并在无定形态S0.94Se0.06/C 中形成了S—Se 键。Se 掺杂与多孔碳结构的协同作用使电池在碳酸酯类电解液中具有长期稳定的充放电循环性能,其中S0.94Se0.06/C复合材料在1 A/g、500次循环后的比容量为910 mAh/g,在0.2 A/g、100次循环后比容量为1105 mAh/g,在20 A/g 时的良好倍率性能为617 mAh/g。此外,在充放电循环过程中没有检测到中间相,并形成了相对稳定的正极CEI膜,说明S1-xSex/C 复合正极成功实现了固固转化。Zeng 等人[58]的工作中也得出了类似的结论,他们将S1-xSex(x≤0.1)渗透到多孔碳纳米纤维(PCNFs)中制成柔性电极,其中S0.94Se0.06@PCNFs 在酯类电解液中5 A/g的高电流密度下,2000次循环后比容量为527 mAh/g,每次的容量衰减率仅有0.026%(图8)。此外,该课题组发现在S0.6Se0.4@CNFs 薄膜正极材料中也出现了相同的现象。单一放电平台的实现归因于S0.6Se0.4复合材料与CNFs 基体之间形成的C—S 键。上述两个工作都指出了在正极上形成CEI 层的理论合理性[59]。碲(Te)是另一种重要的硫族元素,通过Te—S键可以实现与硫无限混合。Sun 等人[60]将TexS1-x分子限制在有序介孔碳CMK-3中,TexS1-x/CMK-3 正极表面会形成CEI 膜阻止多硫化物与碳酸酯类溶剂反应。
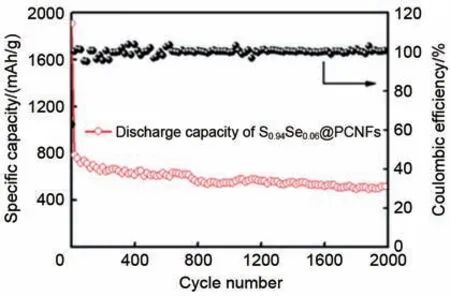
图8 以S0.94Se0.06@PCNFs为正极的Li-S电池在5 A/g的第2000次循环[58]Fig.8 Long-term cycling performance of S0.94Se0.06@PCNFs electrode in Li-S batteries at 5 A/g for 2000 cycles[58]
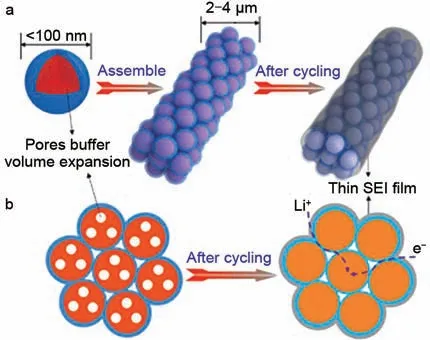
除同族元素外,磷也被证实是改变反应路径的有效元素。Lin等人[61]报道的多硫磷酸锂[Li3PS4+n(0 图9 S-KB-P2S5复合电极的硫氧化还原机理示意图[62]Fig.9 Schematic diagram of the sulfur redox mechanism of S-KB-P2S5 composite electrode[62] 一些过渡金属,如Mo、Fe、Ti、Nb,可以与硫结合形成金属硫化物,表现出独特的充放电机制。Ye等人[63]首先提出了“硫当量正极材料”的概念,即指具有与单质硫相对应的2 V 的高工作电压,以及与传统硫正极相匹敌的高硫含量(>40%)的材料。用MoS3代替单质硫作为正极材料,可以在碳酸酯类电解液中长期稳定循环,紫外-可见光谱(UV-vis)和X射线吸收光谱(XAS)分析表明,在电池充放电循环过程中,不存在多硫化物中间体,在脱锂化过程中,MoS3的非晶链结构没有受到影响。Li等人[64]引入了Fe元素作为中间连接体与硫原子结合,将循环过程中长链Li2Sx中间体转化为LiyFeSx中间体(图10)。非晶a-FeSx/C (x=2,4,6)正极的容量随含硫量的增加而增大,在碳酸酯类电解液中0.1 A/g 电流密度下500 次的稳定循环后比容量达931 mAh/g;并且在未添加LiNO3的醚类电解液中也具有良好的循环性能。 图10 非晶a-FeSx和LiyFeSx的结构示意图[64]Fig.10 Schematic diagram of structures of LiyFeSx and amorphous a-FeSx[64] TiS4也具有类似的反应过程,Sakuda等人[65]报道了TiS4的结构随锂嵌入/脱出而变化,并阐明了一种混合反应机制,在电极反应过程中发生了两种结构变化:S—S 二硫键的断裂和形成以及Ti 配位的变化,两种变化同时发生,从而产生充放电高容量。Sakuda等人[66]报道了新型a-NbSx(x=3、4、5)正极材料,它结合了单质硫和金属硫化物的优点,可以在酯类电解质中可逆循环。 硫与无机原子的共价键结合引发正极的固固转化反应,该过程通常伴随着充放电时正极CEI膜的形成,整个氧化还原反应过程中没有可溶性多硫化物的溶解穿梭。其中,硫和无机原子的不同比例是影响Li-S循环性能的因素。电极中掺杂原子含量越高,循环稳定性和速率越好,但会影响电池整体能量密度,需要平衡二者达到最优的电池性能。此外,目前仍需要进一步深入研究电池循环过程中复合正极的结构演变。 将硫与有机聚合物主链的化学结合和掺杂杂原子的策略结合起来,通过合理的设计可以实现协同作用。据报道,Se 或Te 等杂原子掺杂是改善硫化有机聚合物正极循环性能的有效途径。 2014年,Wang等[67]设计了硫化硒/碳化聚丙烯腈复合材料作为正极材料,有机聚合物骨架和Se的引入都有助于提高硫正极的导电性。此外,电极表面形成的CEI膜可以阻止可溶性多硫化物与电解液反应。因此,SeS0.7/CPAN正极在1200次循环后比容量达780 mAh/g。由于Se 的含量高于S,对比容量的提高有所限制,因此在后续的研究中需要进一步调整S/Se比例。Li等人[68]制备了具有多通道结构的pPAN/SeS2材料,在0.2 A/g 电流密度下,比容量为1100 mAh/g,在4.0 A/g 电流密度下具有高达2000 次的稳定循环寿命。此外该复合材料中活性物质含量高达63%,有效地提高了硫的负载量。尽管其具有优良的长期循环稳定性,但复杂的合成条件限制了其广泛的应用。Jiang等[69]报道了一种一步加热法制备硫化硒/聚丙烯腈复合材料的方法,S和Se的原子比取决于初始投料比、煅烧温度和时间。利用一步加热合成的材料如S0.87Se0.13/CPAN[70]和Se0.38S0.62@pPAN[71]均具有良好的电化学性能,高硫负载量和贫电解质可以进一步提高其电化学性能。Chen 等人[72]通过在硫化聚丙烯腈中引入了少量Se合成SexSPAN复合材料,其合成的Se0.06S@pPAN正极由于少量Se 掺杂的催化作用使锂离子能够更加快速地扩散(图11),从而实现可溶性多硫化物到固体Li2S的快速转化,阻止多硫化物中间体在醚类电解液中的溶解。 图11 硒掺杂提高多硫化物的氧化还原转化率和反应动力学示意图[72]Fig.11 Schematic diagram of selenium doping improves redox conversion and reaction kinetics of polysulfides[72] Li 等人[73]报道了Te 在S@pPAN 正极中作为共晶促进剂的作用,在充放电过程中可以促进锂离子的扩散,降低其扩散势垒,从而显著增强了反应动力学,避免了多硫化物的溶解。He 等人[74]设计了一种以纳米硫为芯、掺硒硫化聚丙烯腈(PAN/S7Se)为壳的新型硫正极结构(图12)。该正极不仅能在高含硫量(高达68%)的条件下实现固相转化,而且在相对低的E/S比条件下也能实现高容量和稳定的循环性能。 图12 S@PAN/S7Se正极结构示意图。(a)三维视图和(b)电化学循环前后复合正极的简化二维截面图[74]Fig.12 the structure of S@PAN/S7Se cathode.(a)Three-dimensional view;(b)simplified two-dimensional cross-sectional view of composite cathode before and after electrochemical cycles[74] 将Se/Te 元素嵌入到S@pPAN 复合材料可以提高硫利用率和能量密度,但是杂原子/硫的物质的量之比会影响反应动力学和电池容量。未来还需要开发更多种类的无机原子嵌入有机硫共聚物,并进一步研究其对反应机理的协同作用。 电解质是影响氧化还原过程的重要因素。用固体电解质代替液体电解液可以消除多硫化物在液体电解液中的溶解,从而避免穿梭现象。其中,硫化物基固体电解质具有离子电导率高、力学性能好、界面相容性好、易加工等优点。自从2008年,Kobayashi等人[75]报道了一种thio-LISICON Li3.25Ge0.25P0.75S4固体电解质以来,具有与液体电解液相当的高离子电导率的硫化物基固体电解质得到了迅速发展[76]。 固态锂硫电池中正极复合材料一般是将活性电极材料(硫或硫基成分)与固体电解质(锂离子导电添加剂)和导电碳(电子导电添加剂)混合,从而改善正极结构内的离子-电子通道,其中研究人员最常使用机械球磨法来制备这些复合正极材料,确保各种材料充分混合,从而在复合电极中获得更好的离子和电子导电性[77]。Nagao 等[78]将硫、乙炔黑(AB)和80Li2S-20P2S5玻璃陶瓷固体电解质以球磨的方式混合制备了复合正极材料,首次放电比容量为1500 mAh/g,循环200 次后比容量保持为850 mAh/g。Nagao 等人[79]进一步探究了球磨温度对电化学性能的影响,在155 ℃高温下机械球磨了S-AB-80Li2S-20P2S5复合正极材料,其中硫含量为50%的S-AB-SE/80Li2S-20P2S5/Li-In 电池在循环50 次后的可逆比容量为1050 mAh/g,容量保持率为97%。 与机械研磨法相比,气相混合法可以获得更小的颗粒粒径,还可以增强固体电解质和活性材料颗粒之间的紧密接触,从而提高电池的循环性能和速率性能。Nagao等人[80]通过气相处理将硫沉积在导电AB 上,然后与thio-LISICON Li3.25Ge0.25P0.75S4固体电解质进行球磨混合制备了一种复合正极。所制备的复合正极具有较小的硫粒径(1~10 nm),在0.13 mA/cm2的工作电流下循环10次后的可逆比容量为590 mAh/g。Zhang 等人[81]也通过在碳纳米管表面沉积硫制备CNTs@S 材料,并进一步与Li10GeP2S12电解质和AB 混合作为正极材料,柔性碳纳米管的引入可以减轻应力应变和界面电阻,促进反应动力学。 通过引入掺杂剂和控制其比例来调节固体电解质的组成是提高固体电解质的电化学性能的常见策略,元素掺杂不仅可以改善电极/电解质界面,还可以拓宽锂离子的传输通道,从而显著改善电化学性能。在Xu 等人[82]的研究中,发现掺MoS2的Li2S-P2S5(Li7P2.9S10.85Mo0.01)固体电解质具有高达4.8×10-3S/cm的离子电导率和5 V(vs.Li/Li+)的电化学窗口。含该固体电解质混合球磨制备的复合正极可产生1020 mAh/g 的比容量,与使用Li7P3S11固体电解质的复合正极775 mAh/g的比容量相比,电化学性能有所提升,循环稳定性也有所提高。在Xu 等人[83]的另一项研究中,使用了一种新型锂超电子导体Li7P2.9Mn0.1S10.7I0.3固体电解质,具有5.6×10-3S/cm 的高锂离子电导率,高达5 V(vs. Li/Li+)的宽电化学稳定性。研究表明,与液态锂硫电池相比,该S-C-Li7P2.9Mn0.1S10.7I0.3复合正极(图13)具有796 mAh/g更大的放电容量、更好的倍率性能和更佳的循环稳定性。 图13 S-C-Li7P2.9Mn0.1S10.7I0.3复合正极的制备工艺示意图[83]Fig.13 Schematic diagram of the preparation process of S-C-Li7P2.9Mn0.1S10.7I0.3 composite cathode[83] 在聚合物基和氧化物基固体电解质中,部分固态锂硫电池实现了由S@pPAN 等正极材料衍生的固相转化反应。例如,Fang 等人[16]将聚四氟乙烯(PVDF)涂层的S/C 复合材料作为正极,其中正极反应从多步转变为一步反应,可能原因是多硫化物在PVDF聚合物中的不溶性和不稳定性。此外,在复合氢化物固体电解质中也实现了固相反应。2019 年,Kim 等人[84]报道了一种0.7Li(CB9H10)-0.3Li(CB11H12)的快锂离子导体。其室温离子电导率高达6.7×10-3S/cm,在60 ℃下以5 C 的放电速率和1 C的充电速率循环时,循环100次后可逆容量为1017 mAh/g,库仑效率约为100%。 如今,尽管对固体电解质进行了大量研究,但仍然存在一些问题限制了固态锂硫电池的实际应用。首先,该体系的实际能量密度低于液体电解液体系;其次,固体电解质的脆性与正极材料不相容,正极材料在循环过程中具有显著的体积变化,导致界面接触较差;第三,硫化物固体电解质的一些局限性,如对空气中水分的化学不稳定性。但是单质硫正极与固体电解质的结合为实现“固-固”转化提供了新的技术可行性。需要指出的是,单质硫正极在PEO 基固态电解质中仍存在多硫离子溶解问题。此外,阐明固体电解质在锂硫电池中的反应机理也是未来研究的重点。 与“固-液-固”和“固-固”转化反应机制不同,在“准固态”转化中,单质硫首先转化为短链Li2Sx(x≤4),并进一步被还原为固态Li2S。该反应路线将长链多硫化物中间体的生成降至最低,从而阻止锂硫电池的溶解穿梭反应。 为了避免在充放电过程中长链多硫化物的扩散,可以用短链多硫化物代替单质硫作为正极,Shen 等人[85]开发了一种Li-Li2S4电池,该电池体系的放电过程如图14 所示。实验发现即使在84%的高硫含量和4.4 mL/g的低E/S比的情况下,和传统Li-S电池相比能获得更高的放电容量。通过改变电解液环境也可以控制反应途径,Pang 等人[86]通过在二乙二醇二甲醚体系(G2∶LiTFSI)中设计了一种低E/S比(5µL/mg)的锂硫电池。通过对CEI膜成分的分析结果表明,与G2∶LiTFSI=7∶1相比,G2∶LiTFSI=0.8∶1.0 的电解液比例中,硫正极从传统的溶解-沉淀反应转变为准固态反应,抑制了多硫化物溶解和穿梭,电池显示出更好的容量保持率。这是因为贫电解液具有较低的溶剂活性,能够引发“准固态”转化。通过同步优化正极和电解液可以改变正极反应途径,Wang等人[87]采用微孔(<2 nm)均匀分布的活性碳纤维布(ACFC)的限制作用和贫电解液的条件,实现了一种准固态反应放电途径。微孔的约束作用可以将Li2S4转化为稳定的Li2S2,具有自修复的特征,并能高度可逆地利用微孔的电化学活性位点。 图14 Li-Li2S4电池的放电过程[85]Fig.14 Illustration of the discharge processes in Li-Li2S4 cells[85] 锂硫电池的“准固态”转化反应是众多变量综合作用的协同结果。在硫正极方面,除了电极组成的调节,还可以进一步优化电极结构或硫碳比等以提高电池性能。另外,E/S比的调节不仅可以提高整个电池的能量密度,而且可以缓解电解液与锂负极的反应。因此开发贫电解液与锂金属负极保护相结合等措施进一步提高循环性能是至关重要的。 锂硫电池因其优异的理论能量密度和低成本等优点,是研究者们近些年的研究热点。但目前包括硫正极可溶性多硫化物溶解穿梭、锂枝晶严重等问题,导致其实际能量密度与理论能量密度仍存在较大差距。针对锂硫电池反应机理的研究出现了许多新的方法和理论,“液-固-液”转化反应、“固-固”和“准固态”转化反应相互促进、相互启发。为了促进锂硫电池的最终商业化发展,未来仍需要在提高实际能量密度、长期稳定循环、提高氧化还原动力学等方面进行深入研究。 针对实现高能量密度的目标,在不牺牲硫利用率的情况下增加硫含量和负载量是固固转化硫正极的关键所在,未来构建三维多孔结构可能是高负载硫正极稳定循环的可行策略。3D 多孔硫主体可以提高硫阴极的电子导电性,并且加速可溶性多硫化物向固态最终产物的转化,减少循环过程中正极积累的绝缘硫,从而抑制活性材料损失。 针对实现长期循环稳定的目标,虽然采用固固转化的硫正极可以有效地防止多硫化物的迁移,有利于稳定的循环性能,然而,当正极中硫含量增加到较高水平时,这种转化途径也很难维持较长的使用寿命。未来可以通过保持硫正极结构完整性,以及电解液的优化协同固相转化来延长循环寿命。 针对提高正极氧化还原动力学速率的目标,通过引入导电材料、掺杂硫族杂原子、将硫键合到导电官能团中等方法可以提高正极材料的导电性以及加快锂离子扩散,从而促进硫正极的氧化还原转化。对于液体电解液,加入低极性溶剂改变多硫化物的溶解方式,可以提高其输运性质。对于固态电解质,合理设计电极/电解质界面接触,可以促进锂离子的传输。 通过固固转化反应的硫正极未来前景可期。在目前已有的研究基础上,以有机聚合物固硫为代表的S@pPAN 正极综合性能较优,是最有望大规模产业化的硫正极之一,未来含硫量60%的电池体系0.1 C的倍率下预计可逆容量达到1300 mAh/g(按硫计算),电池单体能量密度可以达到200 Wh/kg,可达到动力电池要求,1 C 倍率80%放电深度(DOD) 5000 次后容量保持率超过80%。在高载量(>4 mAh/cm2)、贫电解液(E/S 比<3 mL/g)等条件下,预期可逆容量可以达到1500 mAh/g(按硫计算),能量密度预计可达400 Wh/kg,硫利用率90%。此外,固固转化反应锂硫电池的正极体积变化与硫含量、正极结构设计有关,例如合理设计可使S@pPAN电极在充放电过程体积变化只有30%;内阻与电解液以及正负极接触界面有关,优化电解液组分、改善SEI和CEI膜的成分是有效的控制内阻变化的途径。 总结来说,本文主要对硫正极固固转化反应进行了简要综述。“固-液-固”、“准固态”和“固-固”转化反应各有特色,未来也有可能通过不同机制之间复合甚至协同,实现优势互补;另外,针对硫正极动力学缓慢特点,硫正极催化可能也是重要的研究方向。
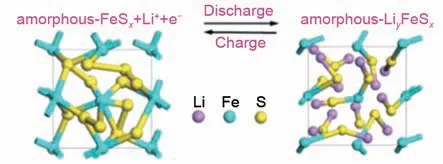
1.4 有机/无机协同固硫

1.5 固体电解质

2 准固态转化

3 结论与展望
猜你喜欢
电池(2022年2期)2022-11-07
电池(2022年4期)2022-11-07
中国建筑金属结构(2019年4期)2019-05-15
哈尔滨理工大学学报(2018年4期)2018-11-24
农业与技术(2018年12期)2018-11-09
中国校外教育(中旬)(2018年9期)2018-09-30
中国美容医学(2017年6期)2018-02-05
绿色科技(2016年20期)2016-12-27
科教导刊·电子版(2016年3期)2016-03-14
农民科技培训(2009年1期)2009-02-17
