
钙钛矿量子点复合光子晶体薄膜的快速制备及结构显色、荧光性能
2022-07-06 13:46李金铎刘志福任瑛琪侯京山刘玉峰房永征
发光学报 2022年6期
李金铎,刘志福,任瑛琪,侯京山,刘玉峰,房永征
(上海应用技术大学材料科学与工程学院,上海 201418)
1 引言
显示在可穿戴系统中起着至关重要的作用,可将人体物理及传感信号进行可视化加工,是诸多光、电、温度信号的输出终端[1]。在以往的研究中,在可穿戴式显示领域有诸多代表性的工作。日本东京大学Someya 教授等利用有机发光二极管制备成一种皮肤显示器,具有低分辨率发光的功能,但目前仍面临诸多挑战,如贴合性、传感特性及阳光下的显色等[2]。韩国KAIST 电气工程学院Choi 教授等利用光纤与有机发光二极管相结合制备出可编织的织物显示器,发现其克服了传统有机管显示在织物中发光性能弱的问题[3]。复旦大学彭教授等利用基于电致发光原理的荧光材料研制了织物基显示系统,成功将显示器件的制备与织物编织过程实现融合,在高分子复合纤维交织点集成多功能微型发光器件[4],但目前该工作仍然需要较高电压的交流电场。尽管国内外学者已做出一定工作,但是采用的技术和原理仍然是传统光电器件与基底的简单嫁接,无法真正实现可穿戴,面显示材料在自然光中的显色和发光弱的问题仍不能有效解决,基础材料端仍然需要更多研发适用性的工作。同时,以OLED 为显示单元的柔性或可穿戴应用面临新的挑战,器件中有机磷光材料发光性能较差,存在随电场越亮越早衰的关键技术难题。诸多研究表明结构光调控是解决显示材料发光强度弱及光调制性能差等问题的重要途径,如新加坡南洋理工大学Hou 等利用电子束刻蚀方法制备了二维光子晶体结构,这种结构可调控钙钛矿的发光强度,并抑制了钙钛矿的自发猝灭[5]。但是物理刻蚀方法技术要求高、成本昂贵,且主要面向低维光子晶体,快速大规模制备三维光子晶体结构仍然是亟需克服的壁垒。
光子晶体是一种在介观尺度下具有周期性自组装结构的材料,特殊的微观结构导致其产生光子带隙现象,同时具有自然光下结构显色的性能[6]。如中国科学院化学研究所宋教授等制备了多种光子晶体胶体球,实现液滴的自组装并研制了多种结构色打印产品[7]。本团队利用光子晶体胶体微球在弧形纤维表面自组装形成结构色纤维[8-10]。同时,研究表明特殊的微结构与发光材料之间存在结构调制的现象,如大连理工大学武教授将CdS 胶体光子晶体与上转换发光颗粒制备成三明治结构,发现它们之间具有光线分离行为[11]。Gan 课题组开展了一种自组装的方法来构建柔性的PCs 来增强CsPbBr3PNC 的荧光,并对机械变形具有灵敏、可逆和可靠的光学响应,为应变计和智能皮肤的开发奠定了基础[12]。光子晶体结构对发光强度和寿命调控是该领域的研究热点之一,但目前构筑光子晶体所用材料较少,只局限于SiO2、PMMA、PS、TiO2等成球型良好的材料[6];组装方法单一,已成熟的组装方法包括重力自组装、垂直沉积法、电泳沉积法[9],导致带隙作用不明显,对发光体的发光调控作用不理想。
无机铯卤化铅钙钛矿量子点是具有低阈值、高量子效率、低成本的新型半导体,在发光显示领域具有潜在的研究价值[13]。但是目前仍然存在环境稳定性差、发光强度低、不能大规模制备等问题。本团队曾利用介孔的微观限域作用制备了性能良好的二氧化硅/钙钛矿量子点,发现其通过微球的隔离可增加稳定性[14]。钙钛矿量子点的发光除了依赖自身的特性外,纳米颗粒周围的环境也对其有很重要的影响,而如果光子晶体结构能形成稳定的微腔,通过费米-金法则可知,珀塞尔因子与态密度成正比,只要匹配光子晶体结构的布拉格散射波长与量子点发光波长位置,即可实现发光性能的增强[15]。因此,本文以二氧化硅(SiO2)胶体球作为光子晶体组装单元,通过电泳沉积的方式制备了厚度可控的光子晶体薄膜。采用热注入方法制备了钙钛矿量子点(CsPbX3,X=Cl,Br,I)。光子晶体薄膜经表面疏水改性后,将钙钛矿量子点嵌入在光子晶体周期性结构中,该结构具有双模显示的效果,具体表现为结构显色和荧光发射。同时,本工作实现了光子晶体结构对钙钛矿量子点荧光效率的增强,在显示、防伪、光电探测器、发光二极管等领域具有潜在的应用价值。
2 实验
2.1 试剂与原料
所有化学品和材料,包括无水乙醇(AR,通用试剂,≥99.7%)、正硅酸四乙酯(AR,通用试剂,SiO2≥28.4%)、氢氧化铵溶液(AR,通用试剂,25%~28%)、氯化铅(99.99%,柯林斯)、碘化铅(99%+,达马斯-β)、溴化铅(99%,达马斯-β)、碳酸铯(99.9%,达马斯-β)、己烷(AR,通用试剂,≥97.0%)、油酸(90%+,阿达玛斯)、油胺(90%+,阿达玛斯)、十八烯(90%,阿拉丁)、六甲基二硅氮烷(99%+,阿拉丁),从探索平台购买,无需进一步纯化。
2.2 CsPbX3(X=Cl,Br,I)钙钛矿纳米晶(PNC)合成
钙钛矿型纳米晶通过热注入方法制备[16]。具体步骤如下:将Cs2CO3(0.814 g,99.9%)、油酸(OA)(2.5 mL,90%)和1-十八烯(ODE)(30 mL,90%)添加到100 mL 三颈烧瓶中,并将混合物在N2下在120 ℃干燥2 h。溶液温度降至100 ℃。将ODE(75 mL,90%)、OA(7.5 mL,90%)、油胺(OAm)(7.5 mL,90%)和PbX2(2.82 mmol)如PbI2(1.26 g,99.99%)、PbBr2(1.035 g,99.99%)、PbCl2(0.675 g,99.99%)装入250 mL 三颈烧瓶中,并在N2下120 ℃干燥2 h。PbX2盐完全溶解后,将温度升高至160 ℃,并快速注入上述制备的Cs 油酸盐溶液(6.0 mL,ODE 中0.125 mol/L)。10 s 后,反应混合物在冰水浴中冷却。对于CsPbCl3合成,添加1 mL 三辛基膦(TOP)(97%)以溶解PbCl2。以7 000 r/min 离心5 min,从溶液中沉淀PNC。离心后,丢弃上清液,将纳米晶体重新分散在无水己烷(99%)中,并通过在3 000 r/min 下进一步离心5 min 再次沉淀,以去除杂质。
2.3 SiO2微球合成
采用优化后Stöber 法合成了SiO2[17]。通过温度和正硅酸四乙酯(TEOS)的用量控制SiO2微球的大小,得到SiO2微球的直径分别为204,237,267 nm,在去离子水中超声分散,备用。
2.4 SiO2 PC 制备
SiO2的光子晶体薄膜是通过电泳沉积[18]的方法制得,即将0.3 g SiO2微球适当分散在20 mL 去离子水中,通过添加几滴NH4OH 将pH 调节至11.2;分别使用ITO 玻璃(20 mm×20 mm)和不锈钢板(25 mm×25 mm)作为工作电极和对电极,在分散液中进行SiO2微球的垂直电泳沉积。将电场保持在4 V,调整沉积时间为3~7 min 制备不同厚度的光子晶体薄膜,并在空气中干燥30 min,备用。
2.5 在SiO2 PC 上组装CsPbX3(X=I,Br,Cl)(PNC)
首先,通过表面改性反应使胶体晶体薄膜具有疏水性,具体方法是将1 mL HMDS(六甲基二硅氮烷)加入20 mL 正己烷中,在45 ℃下放置2 h。其次,将疏水胶体晶体薄膜置于PNCs 的正己烷分散液中渗透组装2 h。
2.6 测试
使用日立UH-4150 光谱仪测定了PNC 的紫外-可见吸收光谱及PC 的反射光谱。PNC 的发射光谱由日立F-7000 荧光分光光度计测得,其中激发的狭缝为5.0 nm,电压为400 V。使用R1 角分辨光谱仪测定了分辨反射光谱、角分辨荧光光谱,使用405 nm 激发器作为激发光源。X 射线衍射数据由布鲁克D8X 射线衍射仪在40 kV 和40 mA条件下,使用Cu-Kα 辐射源测得,扫描范围为5°~80°,扫描速率为6(°)/min。采用双束电子显微镜(FEI helios nanoolab G3 UC,US)获得了SEM 图像。接触角数据由CA-XP1150 型接触角测试仪测得。TEM 数据由日本JEOL 的JEM-2100 型透射电子显微镜测得。
3 结果与讨论
SiO2PC 的局部电场强度分布与PNC 的荧光强度密切相关,光子晶体作为一种电场分布周期性的材料,当光子晶体的入射光产生共振时,电场强度会通过这种共振而增强。当光子晶体的反射光谱峰值与钙钛矿型PNC 的激发波长相匹配,电场强度可实现不同程度增强,从而实现了荧光效率的增强[19-21]。在这项工作中,选择SiO2组装光子晶体,由前期工作可知SiO2携带的表面电荷为负值[8],从而可以通过电泳沉积的方法来实现自组装,并且自组装后形成的光子晶体具有明显的结构色。通过制备204,237,267 nm 三种粒径的SiO2胶体颗粒,用于制备蓝、绿、红三种颜色的光子晶体薄膜。选择了具有荧光发射性能的无机钙钛矿量子点,通过改变卤素种类来改变其荧光峰的位置,实现三色荧光,其与光子晶体带隙的位置相匹配,从而达到光学增强的效果。
图1(a)所示为实验的具体工艺。在垂直电泳的过程中,SiO2微球在电场的作用下会向工作电极移动,SiO2微球在ITO 导电玻璃上逐渐沉积,形成了自组装的光子晶体薄膜。特别地,电泳沉积的特殊工艺使沉积层的厚度可以由电泳沉积的时间来控制。其中电泳沉积过程中,随着电泳时间的增长,在工作电极上沉积的厚度增加,当厚度增加到一定程度时,工作电极表面会被SiO2微球紧紧包裹,类似于一个绝缘体[22];当表面SiO2微球与电解液中的SiO2微球携带相同的电荷,发生同性相斥的形象,将少数已沉积的SiO2微球挤下阳极板,导致厚度减小。图2 为光子晶体薄膜的SEM 图,图2(a)~(e)为电压4 V,电泳沉积的时间为 3~7 min。其中红色数字代表的是光子晶体薄膜的厚度分别为6.64,7.74,10.3,11.7,7.2 μm,在5~6 min,电泳沉积的厚度最厚。图1(b)为SiO2PC 的SEM 图像,SiO2微球具有尺寸均匀的面心立方(Fcc)结构,这是自由能最小的热力学稳定位置,为钙钛矿量子点进入其内部创造了条件。由于SiO2是亲水性,而CsPbX3为疏水性,二者难以实现组装,如何做到使二者相掺杂是我们的关键工艺。CsPbX3的稳定性低于SiO2,所以我们选择对SiO2进行表面修饰处理。使用HMDS 对SiO2进行疏水性处理,其中HMDS 中的疏水基团取代了SiO2的亲水基团羟基,制备出表面具有疏水基团的薄膜。图1(e)显示,SiO2与水原本的接触角是11°,经过疏水性处理后,接触角变为135°(图1(f)),表明SiO2表面的疏水性得到很大的提升,这为CsPbX3进入SiO2的晶格之间创造了条件。CsPbX3的TEM 图像如图1(c)所示,表明PNC 生长良好。得到疏水性SiO2薄膜后,对SiO2薄膜进行CsPbX3组装,得到SiO2/CsPbX3的复合结构(图1(d))。图2(f)为SiO2/CsPbX3的截面SEM,可以清晰地看到CsPbX3进入了SiO2光子晶体内部。

图1 (a)实验流程图;(b)SiO2的SEM 图;(c)CsPbI3的TEM 图;(d)SiO2/CsPbI3的SEM 图;(e)改性前的接触角;(f)改性后的接触角。Fig.1 (a)Experimental flow chart.(b)SEM of SiO2.(c)TEM of CsPbI3.(d)SEM of SiO2/CsPbI3.(e)Contact angle before modification.(f)Contact angle after modification.

图2 (a)~(e)SiO2电泳沉积3~7 min 的SEM 图;(f)CsPbX3进入SiO2晶体间隙的SEM 图。Fig.2 (a)-(e)SEM images of SiO2 electrophoretic deposition for 3-7 min respectively.(f)SEM image shows CsPbX3 entering into the gap of SiO2 crystal.
SiO2在经过电泳沉积后形成了光子晶体薄膜,这些SiO2球体在平行六边形填充(111)平面上密集地填充在Fcc 晶格中,SiO2的折射率是1.45。其中由于SiO2的粒径不同,制备出来的SiO2光子晶体薄膜具有不同的结构色(图3(b)、(f)、(j)),即光子晶体的反射峰是由光子带隙产生,背景波谱被吸收为薄膜中的Fabry-Perot 干涉[23],即两个反射面(空气-薄膜和空气-衬底界面)之间的多次光反射之间的干涉。Fcc 晶格(111)平面的反射峰可以简单地由布拉格定律确定,如下所示:
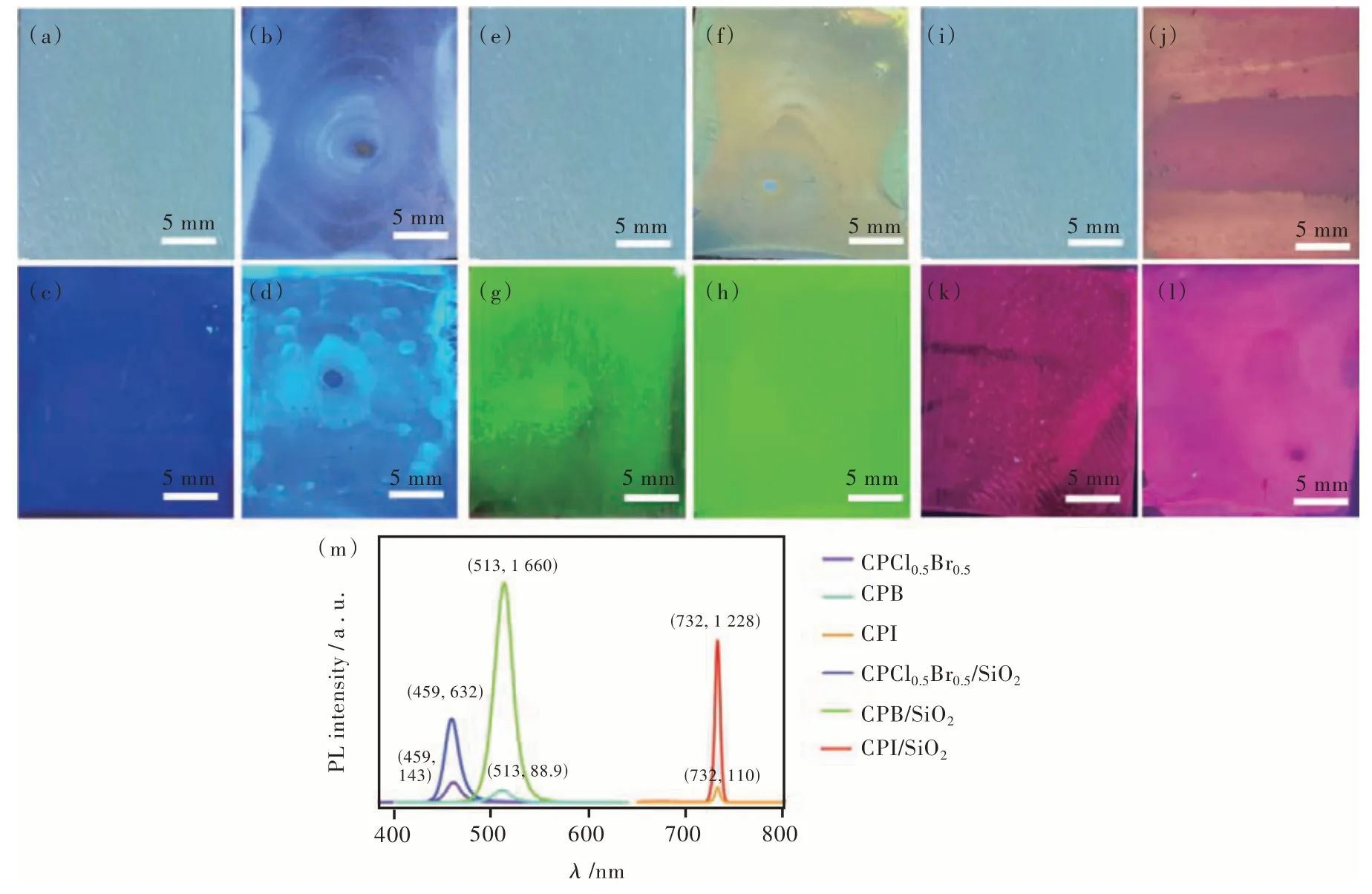
图3 (a)、(e)、(i)玻璃基板上的PNC 在自然光下的数码照片;(b)、(f)、(j)SiO2 PC/PNC 在自然光下的数码照片;(c)、(g)、(k)玻璃基板上的PNC 在365 nm 激发下的数码照片;(d)、(h)、(l)SiO2 PC/PNC 在365 nm 激发下的数码照片;(m)SiO2 PC 对CsPbX 的荧光增强效果。Fig.3 Digital photos of PNC on glass substrate under natural light((a),(e),(i)),PC/PNC under natural light((b),(f),(j)),PNC on glass substrate under 365 nm excitation((c),(g),(k)).(d),(h),(l)Digital photo of SiO2 PC/PNC excited at 365 nm.(m)Fluorescence enhanced effect of SiO2 PC on CsPbX.

其中,neff为有效折射率,np为材料的折射率,nm是空气的折射率,φ是入射角,D为粒子直径。当直径增大时,反射峰逐渐红移。
如图3(c)、(d)、(g)、(h)、(k)、(l)所示,钙钛矿量子点/光子晶体复合薄膜((d)、(h)、(l))具有明显的荧光增强效果,肉眼即可明显地分辨。其中,图3(b)、(f)、(j)是在自然光下PC/PNC 薄膜反射出来的结构色,图3(d)、(h)、(l)是在紫外光下PC/PNC 薄膜发射出来的荧光,薄膜在不同的刺激条件下产生了不同的光响应模式。图3(m)显示了PNC 在玻璃基板上和PC 上的发射荧光光谱,通过荧光强度比值可知,CsPbBr3在PC 上得到了约19 倍的增强效果,CsPbCl3、CsPbI3也分别在PC 上得到了约4.5 倍、11 倍的增强效果。
图4(a)~(c)显示了SiO2PC的反射光谱及CsPbX3PNC的吸收光谱。其中,图4(a)表示的是204 nm 的SiO2PC的反射光谱(蓝色曲线),在460 nm处存在一个反射峰,CsPbCl3PNC 的吸收边(绿色曲线)位于460 nm 左右,与204 nm 的SiO2光子晶体的反射峰很好地匹配;图4(b)是237 nm 的SiO2PC 的反射光谱(绿色曲线),在510 nm 处存在一个反射峰,CsPbBr3PNC 的吸收边(蓝色曲线)位于510 nm 左右,与237 nm 的SiO2光子晶体的反射峰很好地匹配;图4(c)为267 nm 的SiO2PC 的反射光谱(紫色曲线),在700 nm 处存在一个反射峰,CsPbI3PNC 的吸收边(红色曲线)位于700 nm 左右,与267 nm 的SiO2光子晶体的反射峰很好地匹配。当PC 的反射峰和PNC 的吸收边相吻合时,PNC 与PC 发生耦合作用,PNC 的荧光强度实现了成倍数的增强。

图4 (a)~(c)CsPbX3(X=Cl/Br,Br,I)的吸收光谱与SiO2(204,237,267 nm)的反射光谱;(d)~(f)CsPbX3(X=Cl/Br,Br,I)/SiO2(204,237,267 nm)不同镀膜时间的荧光光谱。Fig.4 (a)-(c)Absorption spectra of the CsPbX3(X=Cl/Br,Br,I)and reflected spectra of SiO2(204,237,267 nm).(d)-(f)Fluorescence spectra of CsPbX3(X=Cl/Br,Br,I)/SiO2(204,237,267 nm)at different coating time.
使用电泳沉积工艺的优点之一是可以通过控制电泳沉积的时间来实现光子晶体薄膜厚度的可控化制备。当电泳沉积的电压一定时,时间越久,沉积的厚度越厚。电泳沉积过程采用4 V 的电压,时间是3~7 min,其荧光增强效果如图4(d)~(f)所示,PC 对PNC 的荧光增强效果是先增后减的趋势,CsPbCl3、CsPbBr3、CsPbI3的最佳增强厚度的电泳时间分别为4,6,5 min。这是由于:(1)沉积时间过短时,薄膜的致密度有所欠缺,从而导致电场强度不够均匀,对PNC 的增强效果不佳;(2)当沉积时间太长时,薄膜的厚度较厚。
根据结构的有序程度,光子晶体可分为长程有序结构的有序光子晶体(OPC)和短程有序结构的非晶光子晶体(APC)。OPC 是在不同的视角下表现出明亮的彩虹色结构颜色,APC 通常具有较弱的反射和任何视角时表现出非彩虹色。电泳沉积制备的SiO2光子晶体是长程有序的结构,具有明显的虹彩现象[24-25]。光子晶体的虹彩现象是否对荧光增强效果有影响,是探究的另一个方向。使用了0~360°完整角度范围的角分辨光谱仪(图5(a));采用智能旋转设计,易于控制出射和入射方向;采用智能算法,快速实现反射光谱测量模式;凭借外接的紫外光源,对样品进行角分辨荧光检测[26-27]。以SiO2/CsPbI3为例,SiO2的反射峰在700 nm 左右的,CsPbI3的吸收峰也在700 nm 左右,两者发生耦合作用,CsPbI3的荧光性能得到了很好的增强效果。对267 nm 的SiO2光子晶体做反射角分辨光谱(图5(b)),可以清楚地看出,SiO2光子晶体的反射峰在5°~60°之间不断地发生蓝移并且强度不断减小,这是由SiO2光子晶体的长程有序的结构造成的,具有明显的虹彩现象。SiO2/CsPbI3荧光角分辨光谱如图5(d)、(e)所示,图5(d)表明SiO2/CsPbI3薄膜的荧光强度在0~180 ℃是递减的,图5(e)表明SiO2/CsPbI3薄膜的荧光强度在180~360 ℃是递增的。从图5(d)、(e)可知,SiO2/CsPbI3薄膜的荧光峰在687 nm 处。为了更好地表征荧光强度变化,取SiO2/CsPbI3薄膜的角分辨荧光光谱中687 nm 处做了荧光趋势(图5(f))。结果表明,随着角度的变化,统一峰位置的荧光强度也在变化,这与光子晶体结构的变化趋势是一致的,从而导致二者在匹配的发射峰位置有增强的效果。
众所周知,无机钙钛矿量子点的稳定性较差,在纳米级别的量子点中,晶体容易聚集,其光学性质降低,当量子点应用于器件时,这种现象是降低器件特性和效率的直接因素[28-29]。在器件应用中,如何保证量子点具有高的稳定性和量子效率,一直都是科研的方向[30]。因此,我们设计了一种三明治结构的器件(图6(d)),在原有的发光层上增添了一层PDMS 隔离层,在一定程度上隔绝了氧气和水分,增强了器件的稳定性。如图6(a)所示,4 h 后初始样品的PL 强度比初始值低84%(图6(b))。相比之下,4 h 后添加PDMS 隔绝层的PL强度比初始值低49%(图6(c))。这表明添加PDMS 隔绝层的PL 强度下降明显低于初始样品,表现出显著增强的光稳定性。在这里,T 作为定制的图案,透明的ITO 玻璃作为背景,在自然光下呈现出强烈的红色结构色,在紫外照射下呈现出明显的红色荧光色。当紫外照射去除后,红色的荧光色消失,但结构色再次显现,根据测试结果,该过程是完全可逆的,并且图案在不受到外力的影响下可以保持不变。

图6 (a)PNC/PC/PDMS、PNC/PC 的PL 强度变化趋势(405 nm 激发);(b)PNC/PC 在不同时间的荧光强度衰减;(c)PNC/PC/PDMS 在不同时间的荧光强度衰减;(d)增强稳定性结构示意图;(e)T 在自然光下的数码照片;(f)T 在365 nm 激发下的数码照片。Fig.6 (a)PL intensity variation trend of PNC/PC/PDMS and PNC/PC(405 nm excitation).(b)The attenuation of the fluorescence intensity of PNC/PC at different time.(c)Attenuation of fluorescence intensity of PNC/PC/PDMS at different time.(d)Schematic diagram of enhanced stability structure.(e)Digital photos of T in natural light.(f)Digital photos of T excited at 365 nm.
4 结论
本文研究了光子晶体对钙钛矿量子点荧光强度的增强效果,制备了3 种不同粒径的SiO2微球,并对3 种不同卤素钙钛矿量子点实现了荧光增强。研究发现,蓝、红、绿三色量子点可获得4.5 倍、19 倍、11 倍的荧光增强;同时,利用电泳沉积技术制备了不同厚度的SiO2PC,可实现对PNC 不同程度的增强效果;并且对增强后的PC/PNC 薄膜进行了角分辨性研究,结果表明荧光增强效果呈现先减后增的趋势;最后,通过PDMS 隔离层的保护,使其PL 强度衰减值由51%减小到16%,获得了高稳定性荧光薄膜。
本文专家审稿意见及作者回复内容的下载地址:http://cjl.lightpublishing.cn/thesisDetails#10.37188/CJL.20220079.
猜你喜欢
物理学报(2022年1期)2022-01-19
燃烧科学与技术(2021年5期)2021-10-28
陶瓷学报(2021年2期)2021-07-21
世界最新医学信息文摘(2021年12期)2021-06-09
陶瓷学报(2020年6期)2021-01-26
潍坊学院学报(2020年6期)2020-11-22
装备制造技术(2019年12期)2019-12-25
中成药(2017年6期)2017-06-13
杂草学报(2015年2期)2016-01-04
中国当代医药(2015年29期)2015-03-01

- 发光学报的其它文章
- Preparation of Molecularly Imprinted Polymer Fluorescence Probe Modified by Lanthanide Eu3+Complex and Hemoglobin Sensing Detection
- A Stable UV Photodetector Based on n-ZnS/p-CuSCN Nanofilm with High On/Off Ratio
- Low Boiling-point Solvents Treatment of PEDOT∶PSS Film for Optimized Photovoltaic Cell Performance
- 不同功率O2或N2等离子处理TiNx阳极表面对硅基OLED 发光性能的影响
- 四苯乙烯类聚集诱导发光探针在生物分子检测领域的应用
- 一种比色/荧光增强型碳点基纳米探针用于环境中苯硫酚的高选择性检测