
前列腺癌差异基因标志物筛选有助于预后评价
2022-07-06 03:07陆雪强侯传胜
现代泌尿外科杂志 2022年6期
汪 逊,陆雪强,侯传胜,陈 刚
(复旦大学附属金山医院泌尿外科,上海 201508)
据2020年全球癌症统计报告显示,前列腺癌是男性死亡率第2的癌症[1],目前很难通过环境暴露来完全解释其发生发展[2],已确定的危险因素仅限于高龄、前列腺癌家族史、基因突变(如BRCA1)和疾病(林奇综合征)[1]。流行病学研究已证实,前列腺癌是最具遗传性和最高可抗药性的癌症之一[3-4]。约15%的前列腺癌被诊断为预后不良高风险疾病[5]。在临床上,医生主要通过TNM分期、高危因素以及患者基础情况来判断患者的生存期,但这类指标并不完全准确。而早期诊断和预后预测的困难性往往导致治疗不足或过度治疗,因此,寻找预测前列腺癌不良预后的联合标志物具有重要意义,新的标志物与临床病理学特征以及影像学诊断等结合使用,可有效减少相关风险[6-7]。在本研究中,我们分析了癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库中前列腺癌的原始基因表达数据,以识别参与前列腺癌发生和进展中的关键基因和功能通路;同时结合患者的临床病理资料,深入挖掘这些基因的临床意义,以期寻找相关标志物,更好地判断前列腺癌的发展和预后,为临床医师诊疗决策提供一定的指导。
1 资料与方法
1.1 基因表达谱数据从TCGA数据库(https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga)中下载了498例前列腺癌组织样本及52例癌旁正常前列腺组织样本的基因表达数据以及对应的临床病理资料,数据下载时间为2021年4月。
1.2 数据处理和识别差异表达的基因对TCGA数据,使用TCGAbiolinks包,进行前列腺癌表达数据(Counts/Fpkm)下载和整理,随后利用 DESeq包, 以|FC| >1 &P<0.05作为识别差异表达基因(differential expression genes,DEGs)的阈值,进行肿瘤和正常样本的组间差异分析,得到差异上下调基因。得到所有差异基因后,输出所有差异表达基因的表达矩阵,并用R语言ggplot程序包绘制火山图,将表达上调基因设置为红色,下调基因基因设置为绿色。
1.3 差异基因通路富集分析为确定筛选出的DEGs的功能,用R软件对其进行了基因本体注释(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析。此外,以P<0.05筛选重要通路和功能的临界值设置。GO富集分析采用GOseq包对生物过程(biological process,bp)、分子功能(molecular function,mf)和细胞组分(cellular component,cc)富集的GO term上基因分布情况进行了分析。KEGG Pathway显著性富集分析以KEGG Pathway为单位,应用超几何检验,找出与整个基因组背景相比,在DEGs基因中显著性富集的Pathway。
1.4 KM分析建立基因预后风险模型和临床参数的预后分析用R软件的survival和survimner包,以该基因表达的中位数为二分类变量,采用Kaplan-Meier survival estimate(KM生存分析)以估计筛选出的所有DEGs的生存曲线。以P<0.05作为筛选的临界值。
1.5 临床资料分析对1.4中筛选出的基因,进行临床资料分析。根据临床资料的情况,使用R软件的XML等程序包,对前列腺癌患者clinical T category(cT)、pathologic T category(pT)、pathologic N category(pN)、clinical M category(cM)的各个临床分期分别进行了分析。另外,根据美国癌症协会官网(https://www.cancer.org/cancer/prostate-cancer/detection-diagnosis-staging/staging.html)对前列腺癌最新TNM分期的分类,将患者分为4个Stage,并用Wilcoxon秩和检验对这4个Stage分期分别进行了分析。此外,我们还对筛选出的基因进行了肿瘤和正常样本之间(全部/配对) boxplot图验证。
1.6 风险预测以① 年龄≥60岁、② T3~T4、③ N1、④ M1、⑤ Gleason评分≥8、⑥ 复发作为预后不良高风险因素,以① 年龄<60岁、② T1~T2、③ N0、④ M0、⑤ Gleason评分<8、⑥ 未复发作为低风险因素。分别按照年龄、TNM分期、Gleason评分和复发情况,将前列腺癌患者分成6组不同的高低风险组。DESeq2差异分析寻找筛选出1.4中筛选出的基因的表达情况。
2 结 果
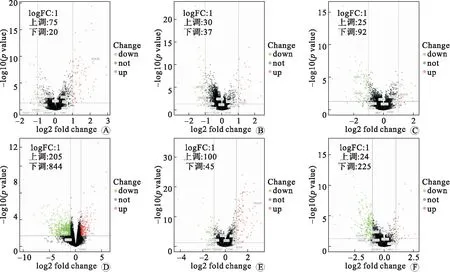
2.1 DEGs识别和富集分析为了获得TCGA数据集差异表达的基因,采用DESeq2包根据|FC| >1和P<0.05的阈值筛选差异表达基因。共筛选出360个差异基因,其中上调基因334个,下调基因26个,火山图显示了来自上调和下调差异表达基因的数量(图1A)。我们通过GO和KEGG通路富集分析,分析了差异基因所参与的生物学功能。根据KEGG 通路富集分析,DEGs主要参与神经活性配体受体相互作用相关通路(图1B)。GO分析结果显示DEGs显著富集在肌肉系统(bp,图1C)、肌动蛋白(mf,图1D)和肌纤维(cc,图1E),三者的综合汇总情况见图1F。
2.2 KM预后分析我们对筛选出的360个DEGs均进行了KM生存分析,以该基因表达的中位数为二分类变量,P<0.05作为筛选的临界值,最终筛选出了12个与OS显著相关的基因,分别是PATE1、TGM4、TPSB2、PRLR、UGT2B17、BCAN、KLHL40、MEI4、CACNG7、CRYGD、OR52E8、OLIG2(图2)。
2.3 TNM分期根据临床资料,对筛选出的12个基因与前列腺癌患者cT、pT、pN和cM的相关性分别进行了分析,结果显示PRLR、BCAN、TPSB2与cT分期相关(P<0.05,图3A~图3C),TPSB2、CRYGD、UGT2B17与pT分期相关(P<0.05,图3D~F),BCAN、CRYGD与pN分期相关(P<0.05,图3G~H),OLIG2与cM分期相关(P<0.05,图3I)。

A:PRLR基因与临床T分期;B:BCAN基因与临床T分期;C:TPSB2基因与临床T分期;D:TPSB2基因与病理T分期;E:CRYGD基因与病理T分期;F:UGT217基因与病理T分期;G:BCAN基因与病理N分期;H:CRYGD基因与病理N分期;I:OLIG2基因与临床M分期。图3 各基因与TNM分期的相关性分析
此外,我们还对前列腺癌患者的临床病理资料进行了整理,将患者分为4个Stage,并用Wilcoxon秩和检验对这4个Stage分期分别进行了分析。结果显示,TPSB2、UGT2B17、BCAN、CRYGD与Stage分期相关(P<0.05,图4)。
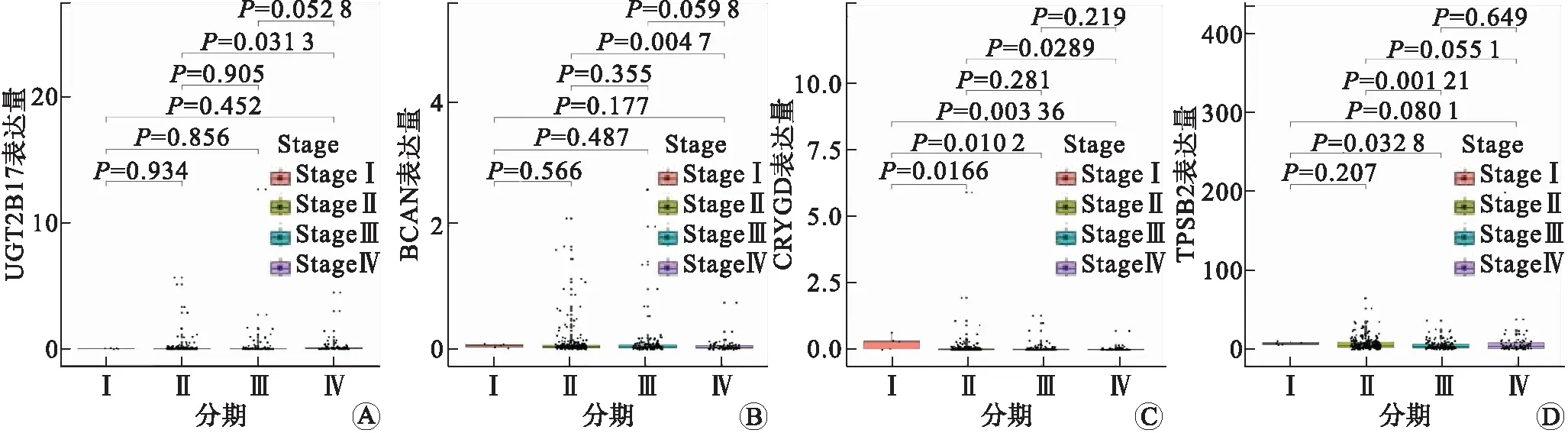
A:UGT2B17;B:BCAN;C:CRYGD;D:TPSB2。图4 各基因与Stage分期的相关性分析
2.4 前列腺癌高低风险组相关性分析分别按照年龄、TNM分期、Gleason评分和复发情况,将前列腺癌患者分成6组不同的预后高低风险组。DESeq2差异分析寻找筛选出的12个基因的表达情况。结果显示,KLHL40在年龄大于等于60组和T3~T4组中表达更多,UGT2B17和OR52E8在年龄小于60组和T1~T2组中表达更多(P<0.05,图5A~B)。KLHL40在N1组中表达更多,PATE1和OR52E8在N0组中表达更多(P<0.05,图5C)。KLHL40在Gleason评分≥8组中表达更多,PATE1在Gleason评分<8组中表达更多(P<0.05,图5E)。KLHL40、PATE1、TGM4、OR52E8在未复发组表达更多(P<0.05,图5F)。
A:年龄;B:T;C:N;D:M;E:Gleason评分;F:复发。红色为表达上调基因,绿色为下调基因。图5 各基因与高低危险组的相关性分析
2.5 样本配对验证我们对筛选出的12个基因的表达量用Wilcox test分别进行了正常和肿瘤样本之间(全部/配对) boxplot图验证(图6)。PATE1、MEI4、CRYGD在正常和肿瘤样本全部和配对boxplot图中P值<0.05。

A、B、C:分别代表CRYGD、MEI4、PATE1基因在肿瘤和正常组织(全部)表达的boxplot图;D、E、F:分别代表CRYGD、MEI4、PATE1基因在肿瘤和正常组织(配对)表达的boxplot图。图6 正常组均和肿瘤样本(全部/配对)表达的boxplot图
3 讨 论
发现可靠度高且能准确预测前列腺癌患者预后的分子生物标志物迫在眉睫。有效的预后生物标志物可提供在没有治疗的情况下特定患者临床预后的重要信息,并弥补现有TNM 分期和高危因素等信息指导临床医生决策的不足,对于治疗方案的选择也是有非常大的参考价值。而各种生物信息学分析技术如单细胞测序[8]、蛋白组学[9]等已广泛用于识别与前列腺癌相关的潜在分子标记物。与之前的研究相比,我们在识别差异表达的基因后,通过KM生存分析筛选了与预后相关的基因,并结合患者的临床病理资料对筛选出的基因与TNM分期和高危因素相关的临床意义进行了深度挖掘。最后还用Wilcox test分别进行了正常和肿瘤样本之间(全部/配对) boxplot图验证。
在筛选出的12个基因中,TPSB2、UGT2B17、BCAN、CRYGD与TNM Stage分期相关。而在高危因素的分析中,KLHL40、UGT2B17和OR52E8与年龄和原位肿瘤大小相关。KLHL40、PATE1和OR52E8与淋巴结转移相关。KLHL40、PATE1与Gleson评分相关。KLHL40、PATE1、TGM4、OR52E8与是否复发相关。最后,PATE1、MEI4、CRYGD在肿瘤和正常样本全部和配对boxplot图中均通过了验证。该部分研究结果已有部分被逐步证实,如UGT2B17在子宫内膜癌中呈现明显的表达上调[10],而且该基因的多态性与前列腺癌易感性有关,其序列的突变会增加罹患前列腺癌的风险[11]。
本研究中仍存在一些局限性:首先,这些生物标志物的大部分研究都是在欧美人群中进行的[12-14],对于中国人群来说,这些标志物是否产生相似的影响,对于预后的判断和评估作用目前尚无法判断,有待进一步研究。其次,我们无法评估从公共数据库获取的数据的可靠性和真实性。这12个基因的预测准确性还需要大规模的独立研究来进一步验证。另外,由于临床资料的限制和缺失,患者还有大量临床参数未进行收集和分析,这可能会导致一些重要的生物学信息被忽略。
总而言之,本研究发现了由12个前列腺癌的预后基因标志物,该组标志物可以作为前列腺癌患者的独立预后因素,并且这一组标志物可作为前列腺癌药物合成的潜在新靶点。此外,根据不同基因标志物及其对应的临床参数,我们可进一步预测患者的不同维度的临床结局,为前列腺癌患者的临床治疗决策提供有效的建议。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
中国典型病例大全(2022年12期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
健康体检与管理(2022年2期)2022-04-15
基础医学与临床(2022年1期)2022-01-21
现代仪器与医疗(2021年6期)2022-01-18
特别健康·下半月(2020年6期)2020-07-06
特别健康·下半月(2019年9期)2019-09-24
晚晴(2017年11期)2017-11-12
健康必读(2016年6期)2016-07-13
