
原发性颅内多发外周T细胞淋巴瘤1例并文献复习
2022-07-05 02:52孙永康丁峰陈三送狄广福程礼敏江晓春
临床神经外科杂志 2022年3期
孙永康,丁峰,陈三送,狄广福,程礼敏,江晓春
原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)是一种少见的非霍奇金淋巴瘤,仅限于神经系统且大部分为B细胞起源,T细胞起源(primary T-cell lymphoma,PTCL)较为罕见。该病发病机制尚不明确,临床表现因发病部位而异,无特异性,诊断比较困难,存在较高的误诊率。回顾性分析皖南医学院第一附属医院2020年7月收治的1例原发性颅内多发外周T细胞淋巴瘤患者的临床资料,结合相关文献,探讨该病的诊断、治疗和预后,提高对该病的认识。
1 资料与方法
1.1 一般资料 患者女,43岁,入院前1个月做家务时,无明显诱因突然出现一过性意识障碍,醒来后感恶心,呕吐数次,呕吐物为胃内容物,休息后缓解。当时未予以重视。后逐渐出现视力减退,家人述其言语不流利,大小便失禁,持续不缓解。既往患有抑郁症10年余。神经系统检查未见明显阳性体征,淋巴结触诊没有异常,胸部、腹部体格检查未见异常。实验室检查未见异常阳性指标。
1.2 影像学检查 头部CT检查示,左侧颞枕叶见片状低密度影,周围边界欠清(图1),建议完善MR检查;MRI检查示,左侧额顶颞枕岛叶、左侧基底节区多发斑片状稍长T1、T2信号影,边界不清,T2-FLAIR呈等高信号,DWI呈等信号,信号不均,增强后不均匀强化,以脑回样强化为主,部分病灶邻近脑沟裂加深,灶周见散在水肿信号影,左侧脑室受压,中线结构向右侧移位(图2)。MRS示,左侧大脑半球多发异常信号灶,病变区NAA峰下降,Cho峰稍升高,Cho/NAA约2.97(图3)。结合患者临床表现及MRI和MRS,考虑大脑恶性肿瘤性病变,炎症性疾病待排除。
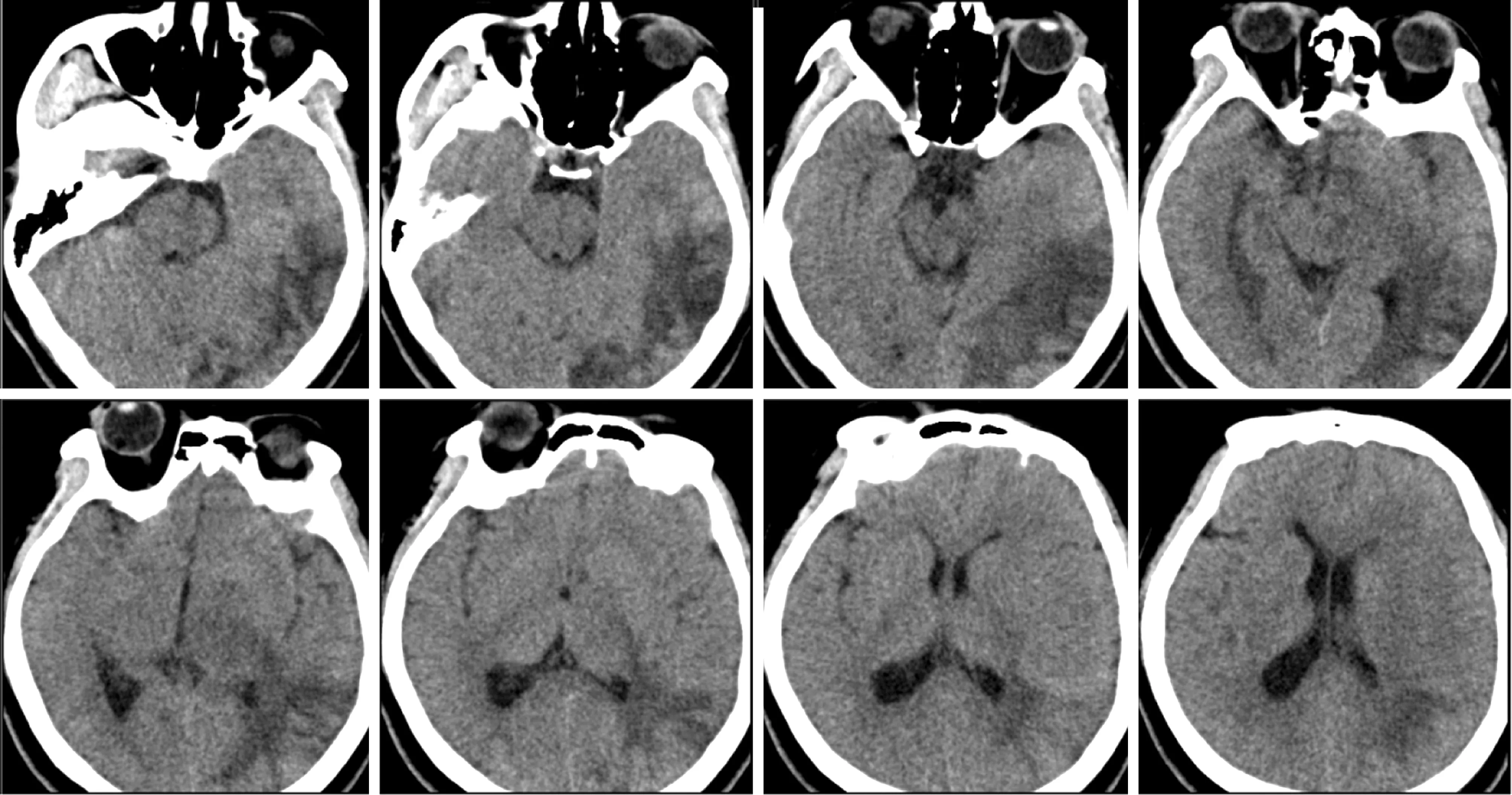
图1 患者头部CT检查结果
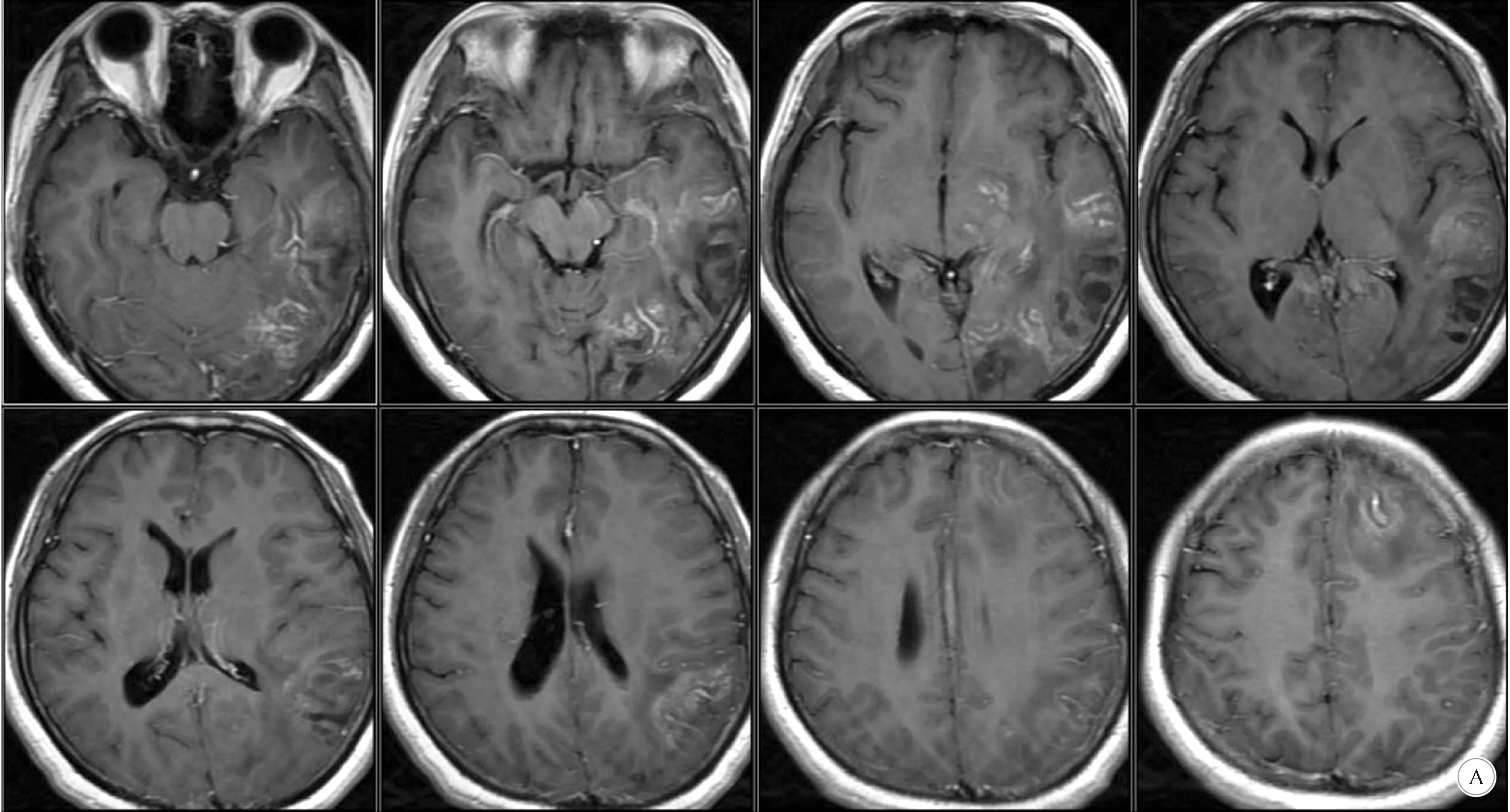
A:T1增强; B:T2-FSE; C:T2-FLAIR

图3 患者MRS结果
1.3 手术方法 患者在完善术前相关检查后,择期在全麻下行左侧额叶炎性病变显微切除术,麻醉成功后,患者取仰卧位,头稍向右偏。标记左额部马蹄形切口,常规消毒铺巾。按标记皮肤逐层切开,皮瓣向下翻,铣开骨窗大小约4.0 cm×5.0 cm,见硬脑膜张力稍高,四周悬吊硬脑膜,X形状切开硬脑膜,见脑压偏高(图4);病变组织呈淡黄色,质地稍硬,与周围组织界限不清,无包膜,血供较丰富;取额上回皮质大小约1.0 cm×1.0 cm×1.0 cm病变组织送常规病理检查,扩大沿病变边缘切除及周边水肿带,见脑压下降,脑搏动良好;术区仔细电凝止血,创面敷以止血纱;清点器械及脑棉无误后,缝合硬脑膜,逐层缝合头皮各层。术毕,术中出血不多,术中麻醉满意,未输血,术后患者恢复自主呼吸后安全返回病房。

图4 患者术中所见
1.4 病理学和分子学检查 镜检脑组织内血管周见淋巴样细胞浸润,呈血管炎样改变,并见散在分布的液化坏死灶,伴胶质细胞增生及组织细胞样细胞反应(图5)。免疫组化标记结果示,CD2,CD3,CD4阳性;CD5个别阳性;CD8,CD20,CD30,CD56阴性;Ki-67高表达。T细胞受体重排检测结果示,TCRB A、TCRB B、TCRB C、TCRG A区段示重排性单克隆峰阳性。综合以上结果考虑该病属于外周T细胞淋巴瘤。
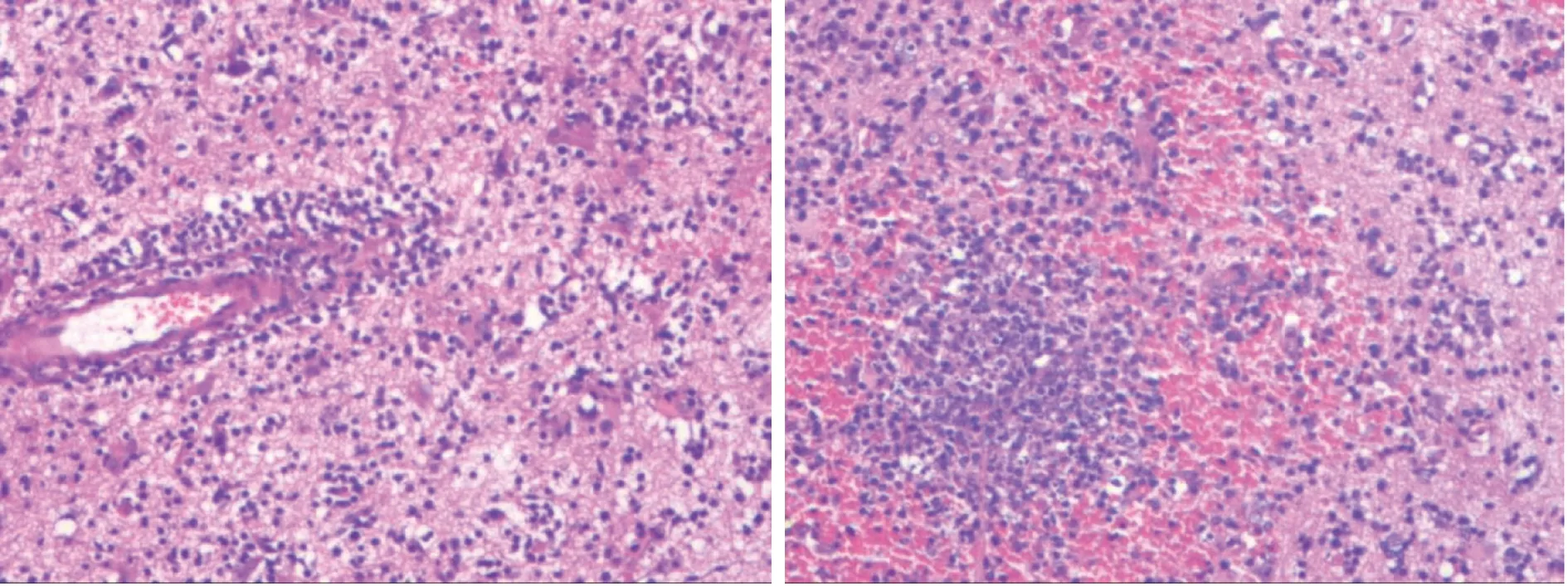
图5 患者病理学检查结果(HE染色,×100)
1.5 随访及预后 患者术后因自身经济条件受限,拒绝接受放疗和化疗方案,本研究尊重其决定,予以签署自动出院书,出院前复查头颅CT示,左侧颞枕叶片状低密度改变,左侧侧脑室前脚受压,中线结构右移(图6)。后予以定期电话随访,其间患者症状逐渐加重,电话随访至第6个月,得知患者病情恶化,在当地医院死亡。
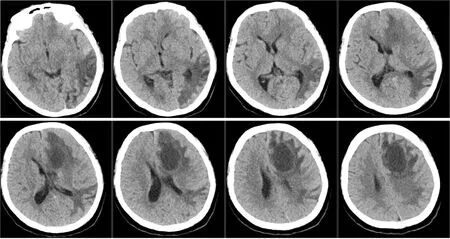
图6 患者术后复查头颅CT结果
2 讨 论
PCNSL是在诊断时没有全身扩散的情况下发生在中枢神经系统(包括眼睛)内的非霍奇金恶性淋巴瘤[1],约占所有原发性脑恶性肿瘤的2%,其中90%以上的PCNSL是B细胞起源[2],弥漫性大B细胞淋巴瘤(diffuse large B cell lymphoma,DLBCL)是其最常见的类型[3],但只有1%~3.6%具有T细胞表型[4]。PTCL在不同的国家,其发病率具有较大差异,有报道显示,在西方国家,PTCL的发病率仅占1%~4%;而在远东国家,该病的发病率高达8.5%~16.7%[5]。该病在男女中均可发生,男性患者发病率略高于女性[6],在免疫功能强和免疫功能低下的患者中都有报道,但其更好发于有免疫功能缺陷的患者[7]。
由于中枢神经系统(central nervous system,CNS)中不存在淋巴组织,因此PCNSL的确切发病机制尚不明确,目前比较流行的有两种假说。第一种假说认为PCNSL来源于外周淋巴细胞的恶性转化,依据是CNS原发和外周发生的淋巴瘤细胞免疫表型并无明显差别。另一个假说认为由于血脑屏障(blood-brain barrier,BBB)的存在,免疫细胞不能进入CNS,颅内环境成为肿瘤逃逸的“庇护所”,从而易于发生PCNSL[8]。然而,淋巴瘤细胞的起源部位,参与淋巴细胞肿瘤转化的生物学机制,以及它们在疾病过程中在中枢神经系统中的限制尚不清楚。 由于其稀有性,T细胞亚型的生物学行为甚至没有得到很好的定义[9]。PTCL的种类很多,最常见的有外周T细胞淋巴瘤( PTCL not otherwise specified,PTCL-NOS)、血管免疫母细胞T细胞淋巴瘤(adult T-cell leukemia/lymphoma,AITL)和间变性T细胞淋巴瘤(anaplastic large T/null-cell lymphoma,ALCL)[10]。
PTCL的临床表现不因世界卫生组织对该病的分类而有所不同,而是由该肿瘤在中枢神经系统生长的部位、侵袭的程度、生长的数量等多种因素共同决定。该病的表现可从细微的行为变化发展到严重的神经功能缺陷,如面部局限性麻木、身体虚弱无力、感觉异常或相应的颅神经疾病[11]。部分患者会出现视力改变的情况,可能是因为病变直接累及枕叶,进而导致颅内压(intracranial pressure,ICP)升高,随后出现视乳头水肿,严重者可影响视觉传导通路进而出现视力障碍。患者出现意识水平的改变可能是由严重升高的ICP、病变累及软脑膜或大的局灶性实质病变引起,有些患者会出现癫痫发作等症状[12]。本研究患者是以突发意识障碍伴视力减退、恶心、呕吐、二便失禁主诉就诊,同时,该患者既往患有抑郁症10年余,在就诊期间,患者的精神症状较前加重。该病的诊断非常具有挑战性,往往是二次诊断,影像学诊断不能很好地确诊,磁共振成像通常显示弥漫性白质疾病,包括双侧大脑半球、脑室周围区、基底节区、丘脑或脑干,并无特异性[13];特别是MRI已被证明,明显低估了疾病的程度[14]。免疫能力强的患者通常只出现一个病变,颅内多个病变在艾滋病患者中最常见[15]。由于PTCL没有特异的临床表现或影像学表现,对肿瘤组织进行活检仍然是诊断的金标准[16]。本研究患者无免疫缺陷,以意识障碍、视力减退、二便失禁等主诉就诊,临床表现不典型,血液学检查无异常,头颅MRI检查显示颅内多发病灶,曾误诊为中枢神经系统炎症并治疗一月余,症状未见好转且上述情况加重。为明确诊断,本研究为该患者设计了一套完整的检查及治疗方案,包括相关的实验室检查。在征得患者及家属同意后,对位于左侧额叶病变进行了手术活检,并进行了病理学检查。然而,组织学和免疫组织学分析并不能足以区分T细胞淋巴瘤和炎症性疾病,PTCL的病理常表现为血管周围淋巴样细胞浸润,呈血管炎样改变,并见散在分布的液化坏死灶,伴胶质细胞增生及组织细胞样细胞反应[17];当存在大量的浆细胞、中性粒细胞或嗜酸性粒细胞时,这些细胞将向有利于炎症过程的方向发展[18],并不能排除急性炎症性脱髓鞘或血管炎等疾病的可能。本研究患者的病理检查结果,与文献记录基本一致。为了进一步明确诊断,本研究继续进行分子遗传学研究,有文献显示,通过检测重排的T细胞(T cell rearrangement,TCR)基因来识别克隆T细胞群体,对于解决这一诊断问题并提供可靠的诊断具有重要意义[19],本研究也对患者的病理标本进行TCR分子学检查,最终确诊为PTCL-NOS。
目前,对于PTCL的治疗,并没有标准的治疗方案,鉴于这些疾病的罕见性并且缺乏大量的随机对照试验,治疗策略常从CNS受累的其他侵袭性淋巴瘤的治疗中借用,如DLBCL。但与DLBCL的治疗结果相比,模仿的治疗方案往往会导致较差的预后[20]。该病通常由手术活检确诊,诊断后,往往进行化疗或放疗。最常见的化疗方案往往是高剂量的甲氨蝶呤, 一项回顾性分析表明,与接受其他非甲氨蝶呤化疗方案的患者相比,高剂量甲氨蝶呤方案可使PTCL患者获得较长的生存期及更好的预后[21]。但是对于甲氨蝶呤的用量,文献中并没有统一的说法。放疗并不单独用于治疗该病,往往结合化疗。该病的预后较差,放疗和化疗可增加该病的预后,中位生存期为10.9~16个月,60岁以下患者的生存率较好。复发或进行性PTCL患者的预后差,中位生存期约为4.5个月[22]。本研究中患者从诊断到死亡仅仅6个月时间。
虽然PTCL在临床上非常少见,存在较高的误诊率和死亡率,但通过回顾该患者完整的诊断和治疗过程,本研究提高了对该病的认识,同时也了解使用多技术进行联合诊断对该病确诊的重要性。本研究患者因经济条件原因放弃接受较完善的放疗和化疗方案,但是为临床积累了的相关诊疗经验,从而为后续的诊断、治疗及预后提供更大的临床价值。
利益冲突:所有作者均声明不存在利益冲突。
猜你喜欢
传染病信息(2022年3期)2022-07-15
健康护理(2022年3期)2022-05-26
新医学(2022年4期)2022-04-23
睿士(2021年5期)2021-05-20
睿士(2020年5期)2020-05-21
睿士(2020年1期)2020-04-03
睿士(2019年9期)2019-09-10
文萃报·周二版(2019年26期)2019-09-10
时代英语·高二(2017年4期)2017-08-11
饮食与健康·下旬刊(2017年4期)2017-05-26
