
ELISA法测定水产苗种中孔雀石绿及其代谢物的残留量
2022-07-05 01:39张倩玉魏海英于盟盟牟丹丹
渔业研究 2022年2期
梁 娟,张倩玉,魏海英,于盟盟,牟丹丹,王 磊
(日照市海洋与渔业研究院,山东 日照 276826)
孔雀石绿(Malachite green,MG)是一种人工合成的有机化合物,它既是染料,也是杀菌剂[1],在治理鱼卵中霉菌和杀灭鱼体寄生虫以及防治鱼类水霉病、烂鳃病等方面效果显著[2-3],也曾被广泛应用于虾卵、蟹卵中寄生虫和真菌的治疗与预防。孔雀石绿进入生物体后,绝大部分通过生物转化代谢为脂溶性隐色孔雀石绿(Leucomalachite green,LMG)并能够稳定地残留在鱼类等水产品的脂肪组织中[4]。经研究证明,MG/LMG具有致癌、致畸和致突变等毒副作用[1-5],已被大多数国家明令禁止在人类可食用的动物源性食品中使用。《中华人民共和国农业农村部公告 第250号》文件中规定,MG/LMG在所有食品动物中禁止检出。但由于MG/LMG价格低廉、疗效好,相关理想的替代药品尚未出现,导致在水产育苗、养殖中其被非法使用的情况屡禁不止,因此通过对苗种中MG/LMG的检测,可以从水产养殖源头上监控水产品的质量安全。
目前,水产品中MG/LMG的检测方法有GB/T 19857—2005、GB/T 20361—2006、SN/T 1768—2006、SC/T 3021—2004等标准方法,但截至目前,针对水产苗种中MG/LMG残留量的检测,国家相关部门尚未发布国家标准、行业标准等标准方法。2007年10月9日,山东省质量技术监督局发布地方标准DB37/T 713—2007《水产苗种鱼药残留限量》,其中规定的方法是大型仪器分析方法——高效液相色谱法。大型仪器分析方法虽然灵敏度高、准确性好,常作为MG/LMG残留检测的确认方法,然而水产苗种的繁育周期短、季节性强,若抽检样品用大型精密仪器进行检测,受限于检测技术复杂、前处理过程耗时长、检测结果滞后等原因,当阳性样品检测结果上报给渔业主管部门时,苗种已被销售。这种情况不但贻误最佳执法时机,而且容易使含有MG/LMG的阳性样品流向养殖环节,从源头就出现水产品质量安全隐患。因此,在实际检测工作中,建立灵敏度高、特异性强、快速简便的ELISA法具有重要意义,可以为渔业质量安全主管部门的管理工作提供及时的技术支撑,提高执法工作的时效性。
目前检测水产苗种中MG/LMG残留量的主要方法有气相色谱-质谱法(GC-MS)[6]、高效液相色谱法(HPLC)[7-8]、高效液相色谱-荧光检测法(HPLC-FLD)[9]、高效液相色谱-质谱法(HPLC-MS)[10]、高效液相色谱—串联质谱法(HPLC-MS/MS)[4,11-12]、超高效液相色谱-串联质谱法(UPLC-MS/MS)[3,13-15]、免疫胶体金法(GICT)[5]等。本文参照GB/T 19857—2005[16]《水产品中孔雀石绿和结晶紫残留量的测定》对美国柏尔生物技术公司提供的试剂盒使用方法进行优化改进,以期建立对不同基质水产苗种中MG/LMG残留量进行定性、定量测定的ELISA分析方法,为基层检测人员降低检测结果的假阳性率和假阴性率以及提高其可靠性提供建议。
1 材料与方法
1.1 仪器和试剂
MK3微孔板酶标仪,赛默飞世尔科技(中国)有限公司;微量移液器、多道微量移液器,赛默飞世尔科技(中国)有限公司;PL202-L电子精密天平,梅特勒托利多仪器上海有限公司;TG16-WS2台式高速离心机,湖南湘仪实验仪器开发有限公司;涡旋混合器,美国Talboys公司;KQ-5200DE超声波清洗器,昆山超声仪器有限公司;DC-24水浴氮吹仪,上海安谱实验科技股份有限公司;10D经济型纯水仪,上海摩勒科学仪器有限公司;DNP-9002BS-III电热恒温培养箱,上海新苗医疗器械制造有限公司;BCD-215KS海尔电冰箱,青岛海尔股份有限公司;-10~50℃、10%~90%RH温湿度表,北京康威仪表有限责任公司;JYL-C022九阳料理机,九阳股份有限公司;96孔MG/LMG检测试剂盒,美国柏尔生物技术公司;去离子水;分析纯乙腈,上海国药集团化学试剂有限公司;分析纯正己烷,天津科密欧试剂有限公司。
1.2 样品
试验所用样品为国家北方虾蟹产业技术体系日照站、山东省现代农业刺参产业技术体系日照综合试验站和山东省现代农业鱼类产业技术体系日照东港综合试验站等产业技术体系试验站提供的中国对虾(Penaeuschinensis)、三疣梭子蟹(Portunustrituberculatus)、刺参(Stichopusjaponicus)、牙鲆(Paralichthysolivaceus)等不同基质苗种样品,以及取自日照市辖区内经济技术开发区、东港区、岚山区等各区县风险监测样品。
1.3 方法
1.3.1 试剂配制
取100 μL 10×浓缩氧化剂(Oxidant solution)液加入到900 μL乙腈中,按9∶1比例稀释为1×Oxidant Solution;将提取物A (Concentrate of extraction buffer A)固体浓缩袋用少量纯水超声振荡至完全溶解,然后转移至100 mL容量瓶中,用纯水定容,配制成1×Extraction buffer A concentrate;取1 mL 10×浓缩样品提取液B(Sample extraction buffer B)加入到9 mL纯水中,按9∶1比例稀释为1×Sample extraction buffer B;取10 mL 10×浓缩样品提取液C(Sample extraction buffer C)加入到90 mL纯水中,按9∶1比例稀释为1×Sample extraction buffer C;将正己烷加入纯水,超声振荡混合均匀,制备饱和正己烷;取10 mL 20×浓缩洗液(Wash solution)加入到190 mL纯水中,按1∶19比例配制1×Wash solution;取100 μL 100×MG-生物素耦合物(MG-Biotin conjugate)加入到9.9 mL MG-生物素耦合物稀释液(MG-Biotin conjugate Diluent)中,按1∶99比例配制成10 mL 1×MG-Biotin conjugate工作液;取100 μL 100×辣根过氧化物酶标记的链亲和素(Streptavidin-HRP)加入到9.9 mL辣根过氧化物酶标记的链亲和素稀释液(Streptavidin-HRP diluent)中,按1∶99比例配制成10 mL 1×Streptavidin-HRP工作液。为保证检测结果的准确性和可靠性,以上试剂,除1×Extraction buffer A concentrate可在室温(22.5±2.5)℃下长期保存外,其余试剂均需现用现配。
1.3.2 样品处理
挑出样品中饵料等杂质,用去离子水清洗、沥干,搅碎成糜状,充分混合均匀;准确称取(2.00±0.01)g样品置于25 mL聚丙烯离心管中,依次加入1 mL 1×Concentrate of extraction buffer A、0.4 mL 1×Sample extraction buffer B和6 mL乙腈,超声振荡20 min,在室温(22.5±2.5)℃ 条件下4 000 r/min离心10 min;取10 mL聚丙烯离心管,并在管中加入300 mg(±40 mg)纯化试剂(MG clean up mix);移取2 mL乙腈上清液置于该离心管中,以最大转速涡旋振荡1 min后,在室温(22.5±2.5)℃条件下静置10 min;(22.5±2.5)℃、4 000 r/min离心10 min;移取1 mL上清液置于另一个10 mL聚丙烯离心管中,50~60℃水浴氮气吹干。加入100 μL 1×Oxidant solution,最大转速涡旋振荡1 min;(22.5±2.5)℃、4 000 r/min离心1 min,静置15 min;依次加入400 μL 1×Sample extraction buffer C和650 μL饱和正己烷,最大转速涡旋振荡1 min,(22.5±2.5)℃、4 000 r/min离心5 min;去除上层正己烷,下层溶液供酶标板进行检测。
1.3.3 酶联免疫反应
移取标准溶液或样品上机液90 μL,置于对应的酶标板孔中;再按以上顺序加入30 μL 1×MG-Biotin conjugate,轻轻振荡混匀1 min,用盖板膜盖板后,在室温(22.5±2.5)℃条件下孵育30 min;小心揭开盖板膜,将孔内液体甩干,每孔加入洗涤液250 μL,轻轻振荡混匀1 min,将孔内液体甩干,重复洗板步骤3次,每次间隔10 s,在吸水纸上倒扣、排干,排干后若有气泡,先用洗耳球吹破,再拍干;每孔加入100 μL 1×Streptavidin-HRP,轻轻振荡混匀1 min,用盖板膜盖板后,在室温(22.5±2.5)℃条件下孵育15 min;再按照上述方法洗板3次;每孔加入100 μL底物(TMB substrate),轻轻振荡混匀1 min,用盖板膜盖板后,在室温(22.5±2.5)℃条件下孵育15~20 min;每孔加入100 μL终止液终止反应,轻轻振荡混匀1 min,供上机测定(在上机前30~40 min打开酶标仪,以使仪器状态稳定,并将测试波长设定为450 nm)。
2 结果与分析
2.1 超声振荡试验
选取中国对虾、三疣梭子蟹、刺参、牙鲆4种不同基质的MG/LMG阴性样品,每种基质样品分别称取相同10份,均添加为1.00 μg·kg-1加标水平,每2份为一组,设5个平行组,在固定超声频率为40 kHz的试验条件下,分别超声振荡10、15、20、25、30 min,以考察超声振荡时间对MG/LMG检出水平和样品乳化现象的影响(表1)。试验结果显示,在10~20 min范围内,MG/LMG检出水平随着时间的延长而提高,样品提取液状态良好,未出现混浊和乳化现象;20 min之后,样品提取液开始出现混浊和乳化现象,且MG/LMG检出水平降低,这可能是多次使用正己烷去乳化层引起MG/LMG损失所致。由此可见,超声振荡适用于MG/LMG的提取,且最佳时间为20 min。

表1 超声振荡时间对样品检出水平和乳化现象的影响
2.2 静置时间验证
样品加入乙腈超声振荡后,过快收集容易同时收集含有少量水滴的乙腈[11],使氮吹操作困难,浓缩时间增加;静置时间过长,易产生乳化、乳化层含水量高等不利后续试验进行的情况[15]。本文对65份不同基质样品进行阳性筛选,以验证静置时间对后续试验的影响:相对于静置10 min,静置5 min氮吹时间延长;静置15 min氮吹时间与其相差不大,但有些样品开始出现乳化现象,可能是样品中溶于乙腈的杂质析出所致。由验证结果可知,本方法的最佳静置分层时间为10 min。
2.3 线性范围与检出限
本文采用0、0.050 0、0.150、0.500、1.50、4.50 ng·mL-16个浓度梯度建立标准曲线,相关系数R2=0.997 0,样品稀释倍数为1.5倍,故将0.075 μg·kg-1定为本方法的最低检出限。与国家标准推荐的HPLC法[17]和LC-MS/MS法[16]相比,ELISA法检出限较低、灵敏度较高(表2),在0.050 0~4.50 ng·mL-1水平范围内,标准曲线线性关系良好(表3),可以对样本中MG/LMG的残留定量测定。由此可知,采用ELISA法作为MG/LMG监控技术手段具有可行性。

表2 ELISA法和大型仪器分析法的检出限与定量限

表3 ELISA法和大型仪器分析法的线性范围
2.4 加标回收率与精密度(RSD)
同大型仪器分析法的自动进样方式不同,ELISA法是手动进样。正确把握可调试移液器“吸二打一”的操作手法[18]是减少偶然误差、增加微量进样稳定性和确保RSD的有效方法。对于食品中禁用药物,回收率应在方法检测下限、两倍方法检测下限和十倍方法检测下限进行加标回收率试验[19]。不同种类的样品之间性质差异很大,会导致不同的基质效应,而基质效应对反应的干扰是影响ELISA法检测结果准确性的重要因素。样品中的杂蛋白、脂肪、色素等物质会影响抗体与抗原的结合,降低方法灵敏度与稳定性,甚至导致检测结果的假阳性或假阴性。本方法选取4种不同基质的MG/LMG阴性样品,分别做0.075、0.150、0.750 μg·kg-13个加标水平的检测,每个加标水平设6组重复,3个加标水平回收率均在79.7%~104.5%之间,RSD测定范围为3.3%~7.9%(表4)。相对刺参苗种,三疣梭子蟹、牙鲆、中国对虾等苗种可能分别由于蟹壳、鱼骨、虾壳等不可食部分的影响,使样品空白的检出水平偏高,导致3个加标水平的平均回收率均较低,但4种不同基质样品的回收率和RSD均符合渔业主管部门的质量控制规定。由此可见,本文所建立的方法具有良好的准确度和RSD,适用于不同基质水产苗种中MG/LMG残留量的测定。

表4 不同基质、不同加标水平回收率和RSD(n=6)
2.5 正己烷试验
为证明饱和正己烷的提取效果,分别用正己烷试剂与饱和正己烷做除脂净化试验,中国对虾、三疣梭子蟹、刺参、牙鲆等苗种各称取6份,均添加1.00 μg·kg-1加标水平,方式设3组重复,分别计算平均检出水平。结果显示,对于同一苗种,用饱和正己烷去脂的样品检出浓度较高(表5),因此用纯水对正己烷试剂进行饱和具有必要性。
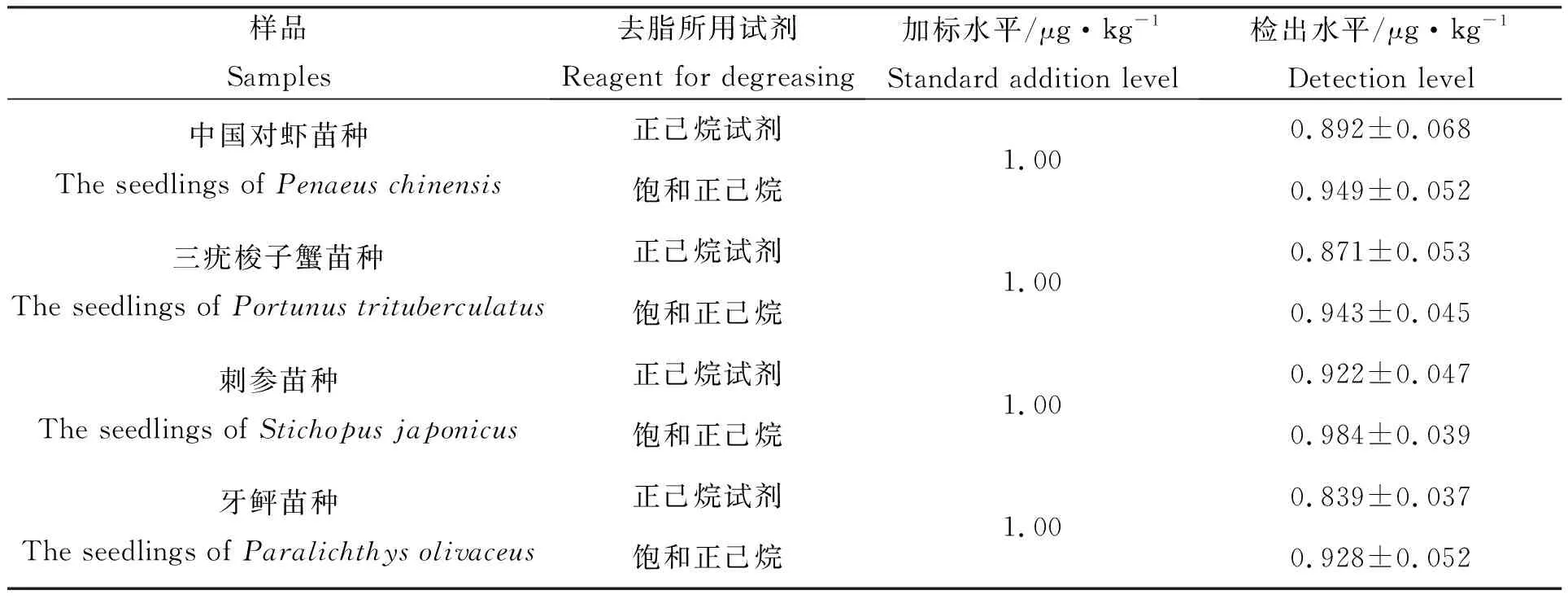
表5 正己烷试剂与饱和正己烷去脂MG/LMG检出水平比对
3 讨论
3.1 提取剂的选择
分析水产品中MG/LMG常用的提取溶剂为乙腈、甲醇、乙醇、戊醇,白志荣等[10]、宋凯等[13]均发现甲醇、乙醇、戊醇等醇类与水的互溶性较强,会将水产品中部分水溶性蛋白提取出来,带来更多杂质,使提取溶液浑浊,不利于后续净化;GB/T 19857—2005[16]《水产品中孔雀石绿和结晶紫残留量的测定》规定提取剂为乙腈;宫向红等[7]研究表明乙腈有利于沉淀蛋白,有一定的净化作用。故本方法选取乙腈为提取溶剂。
3.2 涡旋振荡的优化改进
胡萍等[8]发现采用超声波振荡提取相比涡旋混合器与传统的摇床,更能均质充分与提取完全,可提高反应体系对MG/LMG的结合,同时还有样品反应同步、准确度提高等优点。但是,超声提取时间过短容易混合不匀、提取不充分,造成假阴性;随着超声提取时间的延长,样品中MG/LMG的提取效率有所增加[20],同时也会溶解出更多的杂质成分造成乳化现象。因此,把握好超声振荡时间是操作的关键。在实际检测工作中,一般将20个样品作为同一批次进行检测,每个样品需要涡旋振荡提取4 min,共需80 min。本方法依据GB/T 19857—2005优化为超声振荡提取,该种方式20 min即可完成,使提取时间缩短、工作效率提高,故本方法将涡旋振荡提取优化为超声振荡提取。
3.3 静置时间对样品浓缩的影响
水产育苗用水中藻类较多、水质黏性大,苗种体表清洗不彻底以及MG/LMG具有很强的亲脂性[14]等原因,导致样品加入1×Concentrate of extraction buffer A和1×Sample extraction buffer B振荡混合时容易形成共轭现象,静置时乳化现象严重,分层不明显,不利于MG/LMG的提取。用乙腈作为提取剂,可以减轻乳化现象。对于如何达到更好的防乳化效果,关键应把握好乙腈加入之后的最佳静置分层时间。
3.4 记号笔的优化改进
MG/LMG是常用的三苯甲烷类工业染料,被广泛应用于纺织印染、油墨、涂料中,大部分黑色记号笔油墨中含有MG/LMG[14]。在检测过程中用该颜色记号笔进行标记,会因油墨中MG/LMG的迁移和渗透特性,使样品呈现假阳性。本文对65份不同基质样品进行阳性筛选,筛选过程中离心管用黑色记号笔进行标记,其中有6份样品结果呈现假阳性,假阳率达到9.2%。针对筛选出的假阳性样品,改用红色记号笔对离心管进行标记,用本文建立的ELISA法进行检测,结果为阴性,后续又用LC-MS/MS法对其结果进行确证,结果均为阴性。综上所述,选用红色记号笔对离心管进行标记,可以最大限度地排除黑色记号笔中油墨的干扰,提高检测结果的准确性和可靠性。
3.5 正己烷的优化改进
农业部1192号公告-1-2009《水产苗种违禁药物抽检技术规范》[21]规定,苗种样品的制备应整尾/只绞碎混合均匀。动物源食品中肝脏基质复杂、脂肪含量高,再加上杂蛋白、色素等杂质的干扰,提取液极易产生乳化现象。当乳化现象严重到一次不能彻底去除时,需要增加正己烷的去脂次数。但多次去脂会造成MG的损失,使得样品检出水平降低,容易导致样品的假阴性。本方法将正己烷优化为纯水饱和正己烷,可以在有效去除乳化层的基础上,减少MG的损失,确保方法的灵敏度和检测结果的准确性。
4 结论
本文采取了超声振荡提取、饱和正己烷去乳化层以及用红色记号笔对离心管标记等优化措施,建立了对不同基质水产苗种中MG/LMG残留量进行定性、定量测定的ELISA分析方法。通过检测实际样品进行验证,4种不同基质样品(中国对虾、三疣梭子蟹、刺参、牙鲆等苗种)中MG含量在0.050 0~4.50 ng·mL-1范围内呈现良好的线性关系,相关系数R2=0.997 0,方法检出限为0.075 0 μg·kg-1。在0.075 0、0.150、0.750 μg·kg-1的加标水平下,平均回收率为79.7%~104.5%,RSD为3.3%~7.9%,由此可知优化措施具有可行性。与大型仪器分析方法相比,ELISA法还具备分析成本低、操作步骤简单、有机试剂使用量少、对人体伤害和环境污染危险性小等优点,是目前水产苗种中较为理想的应急筛查和常规检测方法[22]。通过多年实际检测结果可知,用本方法检出的阳性样品,后续再用GC-MS/MS法进行对其确证,检测结果二者一致。因此,本方法可用于实际样品的检测,能够较好地满足水产苗种质量安全监管部门的需要。
猜你喜欢
科教新报(2020年23期)2020-07-21
科教新报(2020年22期)2020-06-11
中国化工贸易·下旬刊(2019年10期)2019-10-21
睿士(2019年11期)2019-08-28
农家科技(2019年1期)2019-03-13
电子技术与软件工程(2016年24期)2017-02-23
中国美容医学(2016年1期)2016-03-17
中国医药科学(2015年4期)2015-05-20
新媒体研究(2014年10期)2014-06-26
中国信息化·学术版(2013年3期)2013-06-25
