
SiCp/Al基复合材料界面结合的第一性原理分析*
2022-06-29 07:26:04杨申欢刘翠霞刘孟宇杨浩邦
西安工业大学学报 2022年3期
杨申欢,刘翠霞,刘孟宇,杨浩邦
(西安工业大学 材料与化工学院,西安 710021)
铝基复合材料的性能优异,尤其是颗粒増强铝基复合材料,具有良好的可再加工性和尺寸稳定性[1],在航空航天、精密仪器、汽车、先进武器等领域中具有广泛的应用。SiCp/Al基复合材料因其具有耐高温、耐磨损、耐腐蚀、高强度和高刚度、热膨胀系数可调等优异性能,作为现阶段理想的新型结构材料之一,在航空航天、军工和民用领域具有广阔的发展前景和巨大的应用潜力[2-3]。
铝基复合材料大规模的发展还相对滞后,其主要的原因是存在界面问题,进而影响界面润湿、决定界面结构。文献[4]通过实验与第一性原理相结合的方法,研究了SiCp/Al基复合材料的界面结构和界面结合等性质。对几种模型的研究表明,界面缺陷会对界面结合产生重大影响,界面的结合强度又直接影响材料的性能。界面的原子构成、原子排列以及键合方式,都不同于界面两侧的基体与增强体,在复合材料服役和变形过程中起着至关重要的决定性作用[5-6]。文献[7]在第一性原理的理论基础下,研究了Al和4H-SiC的界面结合情况,通过计算4H-SiC的Si封端和C封端的成键强度,发现Al-C之间的相互作用强于Al-Si,Al-C结构更稳定,更容易达到平衡状态。金属基体与增强体的界面结合状况直接影响复合材料的性能,作为连接SiCp/Al复合材料增强体与基体之间重要区域,能不能在界面上有效地将施加于基体的载荷传导到增强相上是事关复合材料性能的重要影响因素[8]。
近年来,随着计算技术的快速发展,计算机模拟技术在材料的开发研究中发挥着重要作用,材料的研发模式由传统的实验方式逐步转为理论模拟与实验相结合,指导实验设计的新模式[4]。其中第一性原理的计算方法又是其中最为可靠的,它只依赖几个基本参数就能够计算出体系的能量及电子结构等物理特性,确定材料的结构和基本性质,实现原子尺度的精确研究。文中拟采用以密度泛函理论(Density Functional Theory,DFT)为基础的第一性原理(First-Principles Calculations,FPC)对SiCp/Al复合材料界面结合情况进行研究,建立SiCp/Al基复合材料界面结构,计算形成焓、结合强度、断裂韧性、布居数以及界面成键类型,比较顶位和桥位界面结合情况,以期为进一步研究提供理论基础。
1 几何模型建立
文中以SiCp/Al基复合材料为对象,结合基于密度泛函理论的第一性原理进行研究。建立Al基体和SiCp的晶体结构,如图1所示,粉色球体均为Al原子,黄色球体均为Si原子,灰色球体均为C原子。
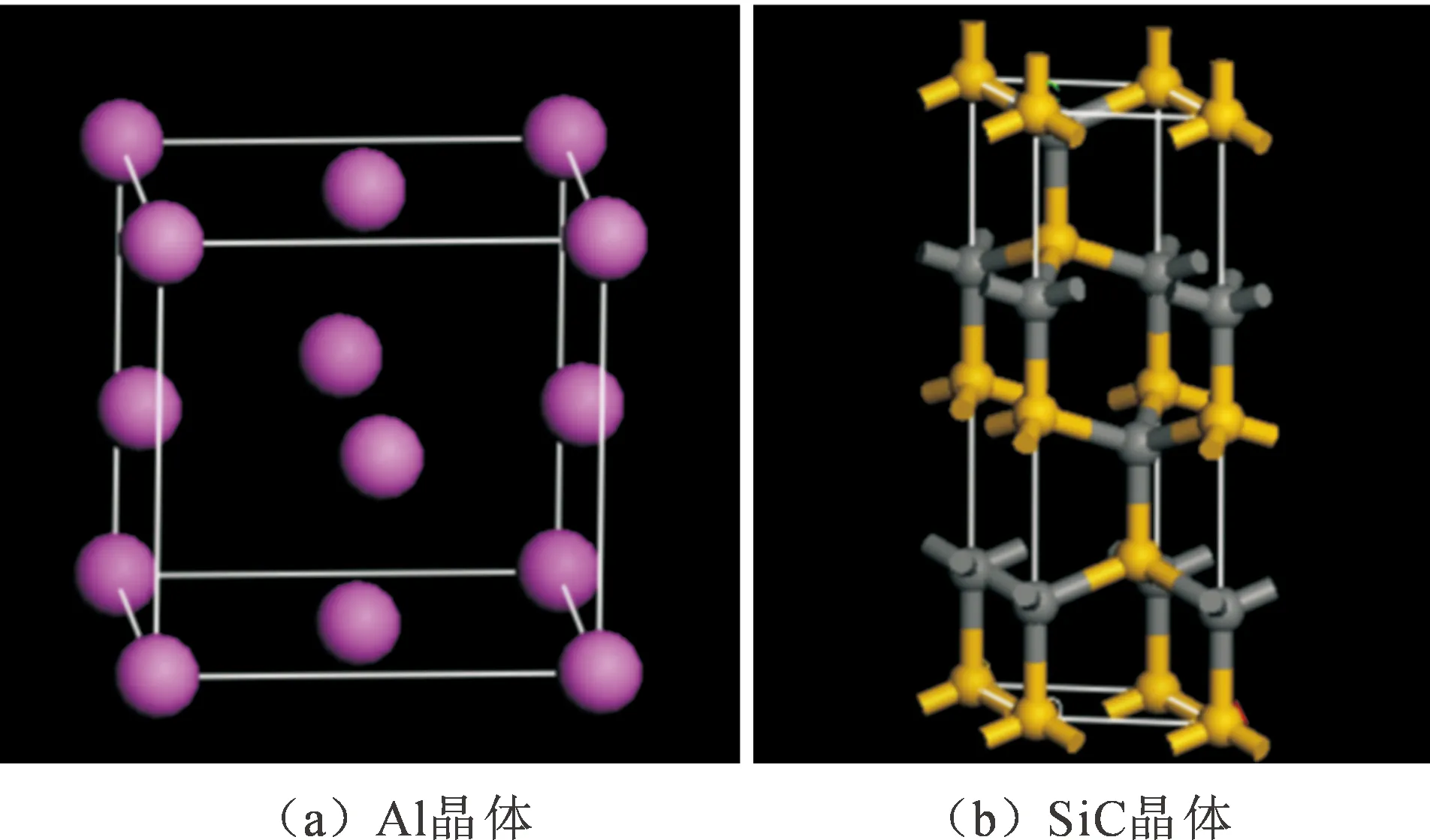
图1 Al和SiCp晶体结构Fig.1 The structures of Al and SiCp crystals
对所建立的体系分别进行结构优化,使其能量达到最稳定的状态,对结构优化后的体系切面,采用包含真空层的建模方法,分别建立Al(100)、Al(110)、Al(111)和SiCp(0001)低指数面的表面模型,如图2所示。
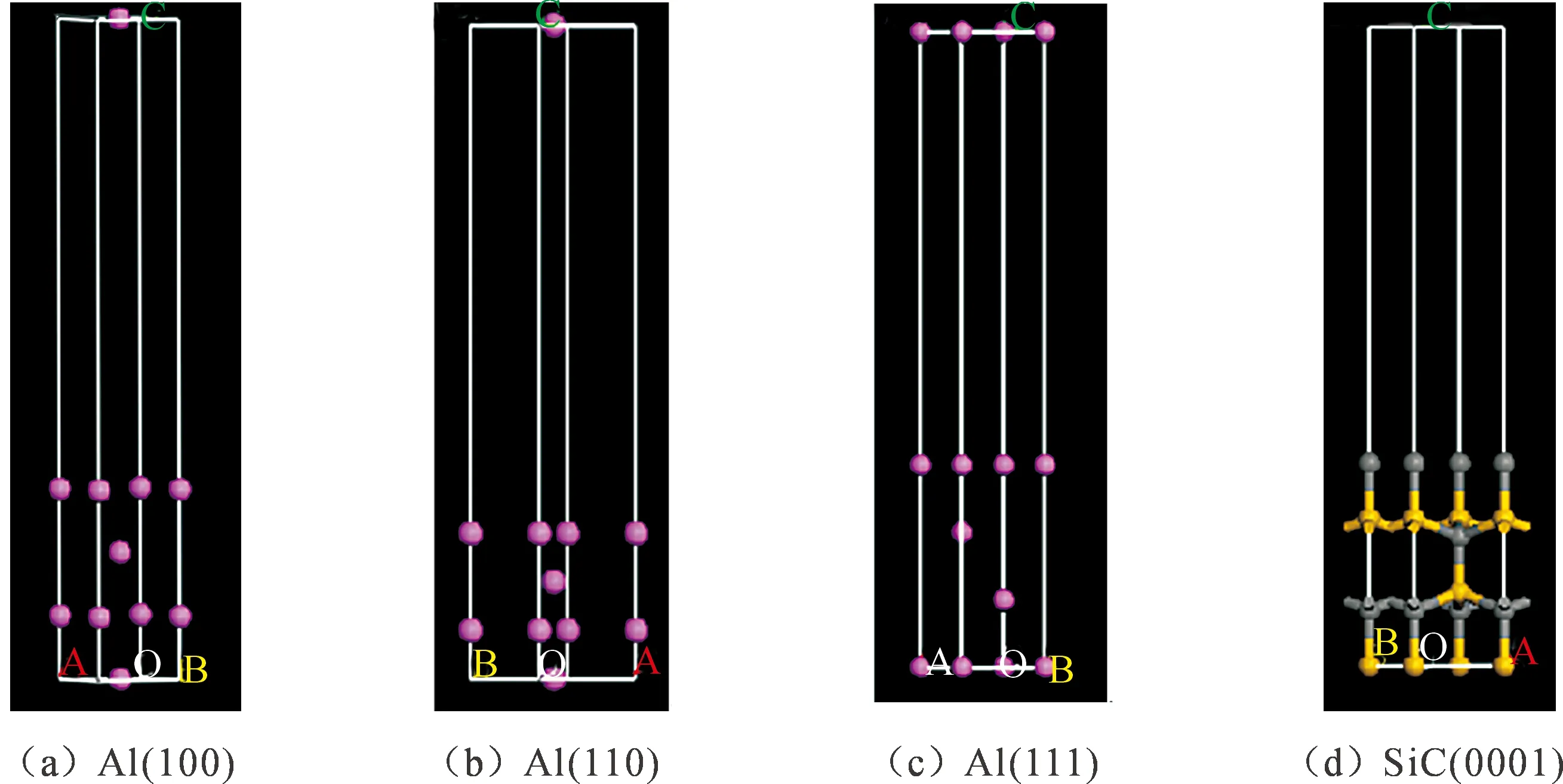
图2 Al和SiCp低指数面的表面模型
将两表面堆垛形成界面结构,根据不同的表面封端,界面原子堆垛位置和体相原子堆垛关系,界面结合分为两大类,分别为Al原子与SiCp中C原子结合的Al-C界面(或称其为C截断面)以及Al原子与SiCp中Si原子结合的Al-Si界面(或称其为Si截断面)。又根据与Al原子结合的原子位置的不同进一步划分为桥位(bridge)和顶位(top)[9-10]。建立的Al-C顶位界面、Al-C桥位界面、Al-Si顶位界面和Al-Si桥位的各个界面结构如图3所示。
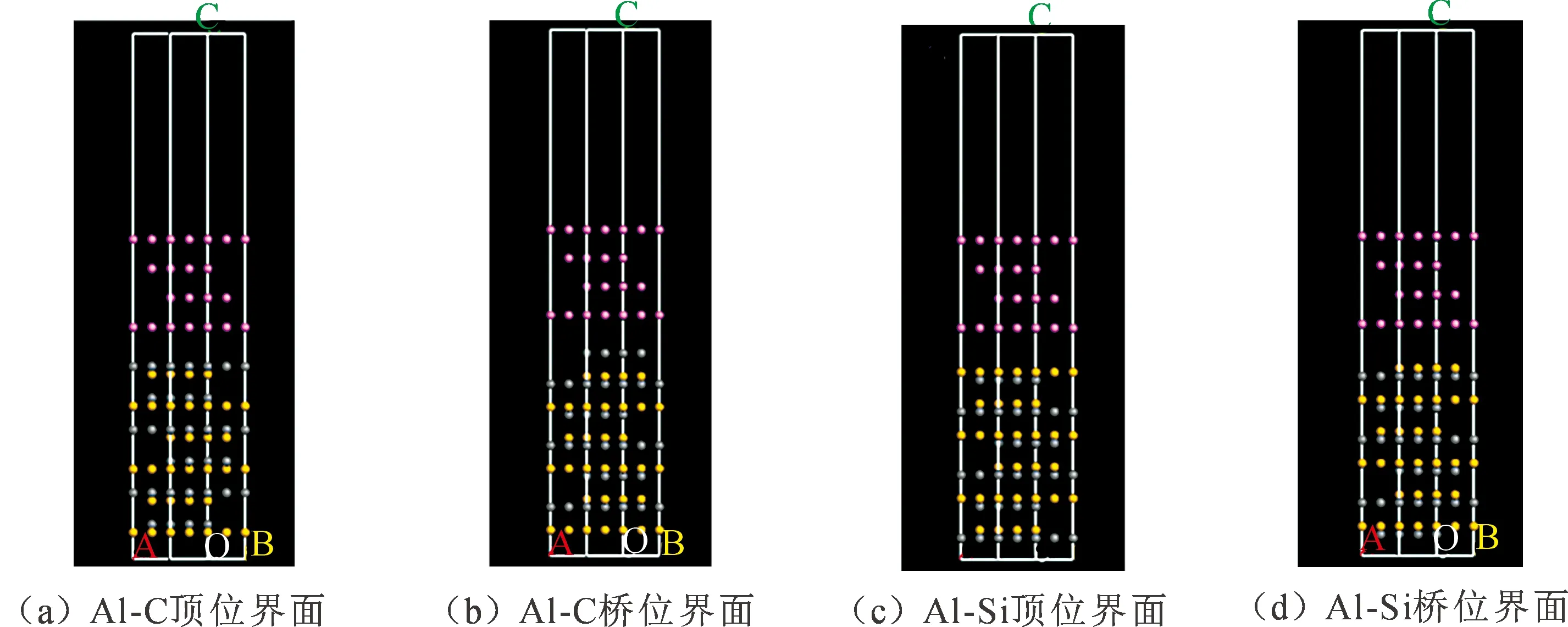
图3 C和Si截断面的界面结构
2 结果与分析
2.1 Al和SiCp各界面表面能
为了研究SiCp/Al复合材料的界面结合性质,需要考虑表面能,确定SiCp/Al基复合材料最稳定的结合界面取向,表面能计算式为
(1)
式中:Esurf为表面能;Eslab为包括真空层在内的整个体系的总能量;Ebulk为一个原子晶胞的总能量,Nslab为整个体系内原子的数目;Nbulk为一个原子晶胞内的原子数目,A为材料表面的面积。结合式(1),计算出的Al、SiCp各低指数面表面能见表1。

表1 Al和SiCp低指数面表面能
由表1中的数据可知,Al(111)晶面具有更低的界面能,即Al(111)界面具有更高的表面稳定性。其原因是Al的(111)晶面是密排面,原子排布密度最大。对于SiCp来说,由于其低指数晶面只有(0001)面需要考虑,故不再计算其他晶面的表面能。
2.2 SiCp/Al基复合材料界面热力学稳定性
众所周知,大多数合金元素进行化合反应都是放热反应,即生成物的总能量比反应物的总能量小,发生反应后,能量差为负值。故反应热的大小可表征晶体结构的稳定性,数值越小,此结构越能稳定存在[11]。在第一性原理的计算中,默认压力是恒定不变的,故体积不受压力影响,则形成焓等于反应热(或形成热)。界面形成焓[12](Formation Enthalpies)ΔEint表示为
ΔEint=EA/B-EA-EB,
(2)
式中:EA/B为界面的总能量;EA为单侧材料A的能量;EB为单侧材料B的能量,界面形成焓的计算结果见表2。

表2 界面形成焓
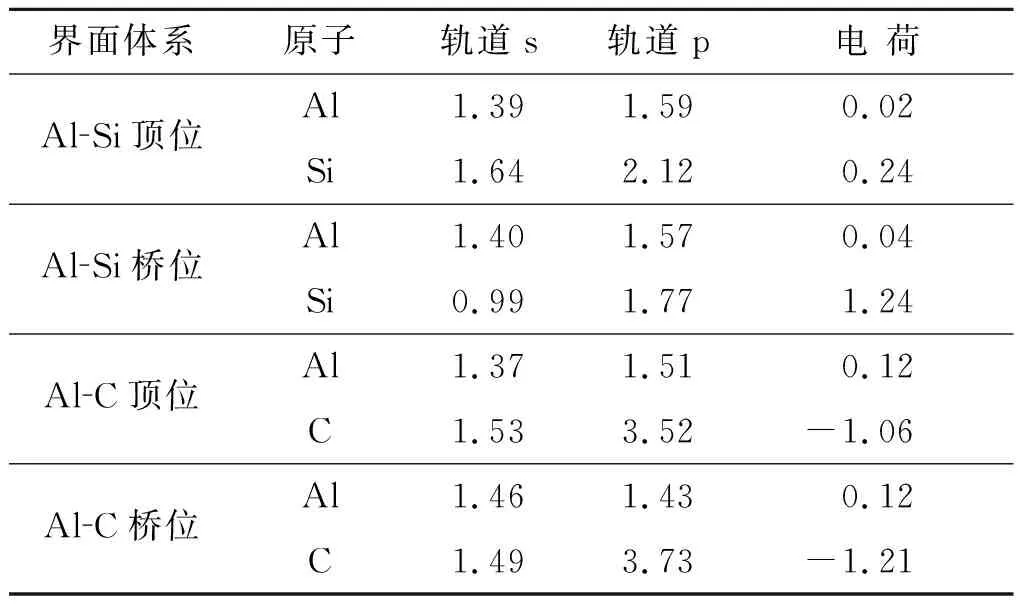
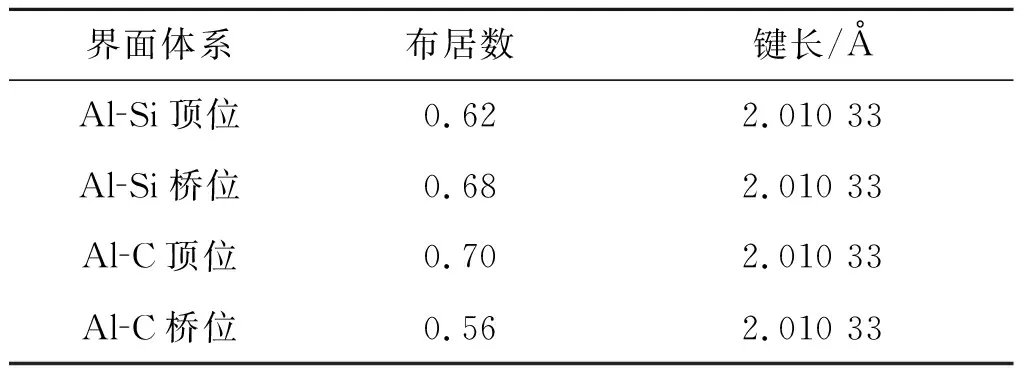
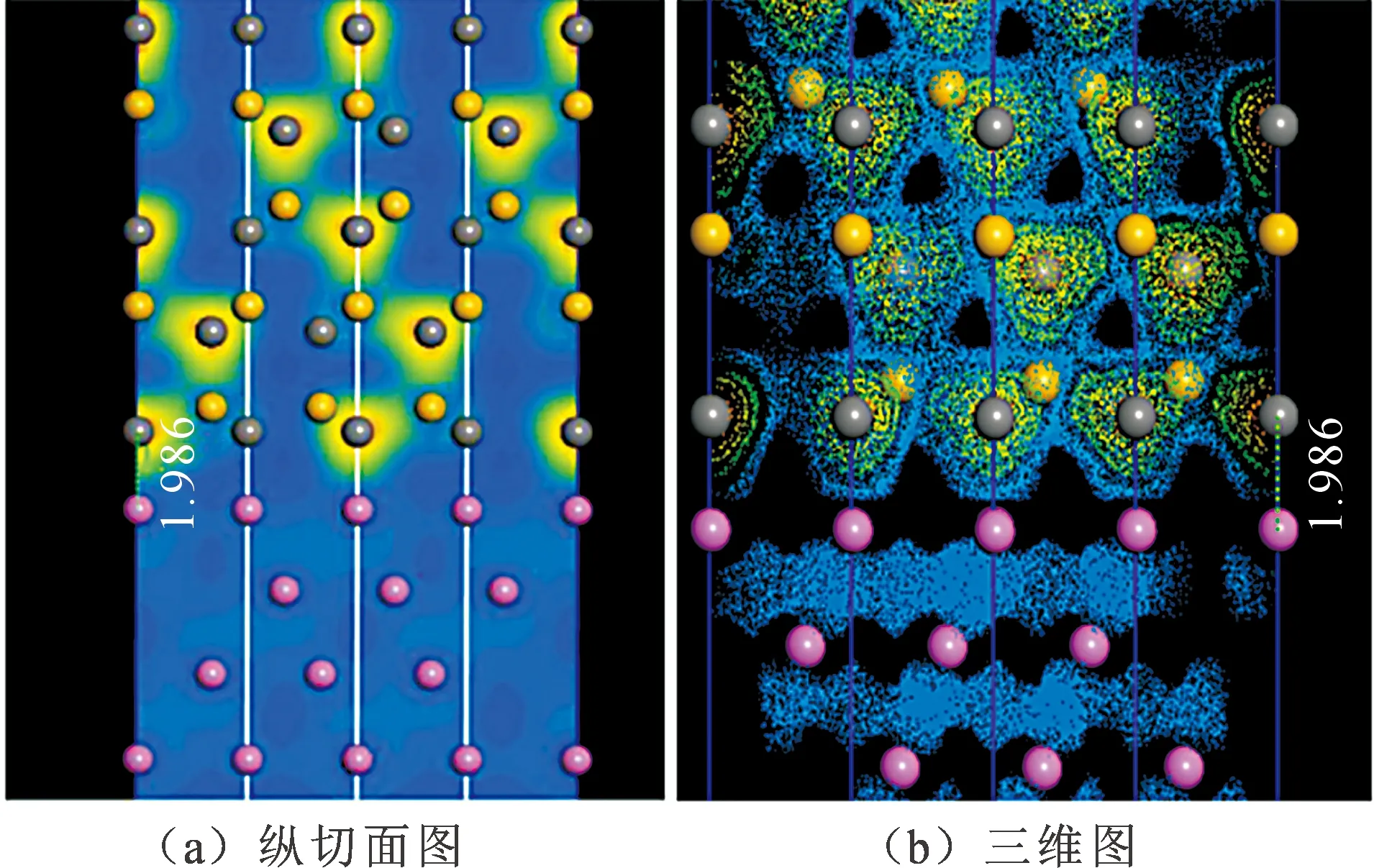
由表2可看出,对于界面形成焓,Al-C顶位 界面强度表示的是两个界面结合的优良程度,如果两个界面不能很好的结合在一起,很容易在界面出发生断裂等一系列缺陷,界面强度的计算式为 (3) 式中:E1和E2为组成界面的两部分各自的能量;Etotal为界面结合后界面体系的总能量。A为界面的面积。 通过式(3)计算相关参数得出表3。 表3 界面结合能 由表3的相关数据我们可以看出,对于顶位位向,Al-C界面比Al-Si界面的结合强度要高;对于桥位位向,Al-Si界面要比Al-C界面的结合强度高;虽然Al-C界面和Al-Si界面顶位位向的结合强度差距较大,但是Al-C界面和Al-Si界面桥位位向的结合强度差距比较小,甚至Al-Si界面桥位位向的强度要比Al-C桥位界面的更强。 从基体到增强相之间的应力传递及转移过程与这二者之间的界面具有直接关系。界面开裂是SiC增强铝基复合材料失效的一个关键原因。从材料科学观点出发,界面断裂失效可以定义为将异质界面分离形成两个不同均匀相的过程,断裂韧性是用以描述界面对裂纹扩展的抵抗能力。Wad为界面结合能,常被用于评价界面断裂韧性和热力学特征[13],Wad与裂纹扩展所需临界应力σF之间的关系可用Griffith[14]方程描述: (4) 式中:E为杨氏模量;c为裂纹长度。可见,Wad越大,则临界应力σF越大,且断裂韧性越高。此外,根据文献[15]的研究,沿着特定[hkl]方向的界面断裂韧性可由下式进行计算: (5) Mulliken布居指电子在各原子轨道上的分布,通过对电荷布居的分析,可以了解界面处原子间的成键情况和电荷转移量[16]。对两种界面结构进行Mulliken布居分析,定量地分析所形成的化学键。原子布居数直观反映各轨道电子占据情况和所携带电荷数;键布居数则表示两原子成键间电子云的重叠程度,反映原子成键强弱,布居数越大说明其成键越强,键长越大,键能越小[17]。 由表4可知,SiC/Al的四种界面结构中,Al和Si原子均失去电子带正电,C原子均得到电子带负电。Al-C之间得失电子数明显大于Al-Si之间得失电子数,电子转移更加明显,并且在Al-C顶位界面中,Al-C之间得失电子数最大,说明Al-C顶位的成键更为稳定。 表4 界面原子布居 表5为界面的键重叠布居,一般情况下,重叠布居数越接近于1,表明共价键越强;越接近于0,表明离子键越强[17]。SiC/Al的四种界面结构中,Al-C顶位界面的布居数最大,为0.70,Al-Si桥位界面次之(0.68),Al-Si顶位界面布居数为0.62,Al-C桥位界面布居数最小,为0.56。这说明Al-C顶位界面的共价键强于其他三个界面结构,同时Al-C之间成键更稳定。这也是Al-C顶位界面的结合能最高,界面更稳定的原因。 表5 界面键重叠布居Tab.5 The mulliken bond population of the interfaces 电荷密度图代表了电荷的聚集程度,图4为Al-C端顶位界面结构的电子分布,可以看到,在SiCp内部每一个C原子与最近的Si原子之间存在一个比较大的电子密度梯度,又由于C的电负性为2.5,Si的电负性为1.8,所以C-Si结合的共用电子对会偏向于C原子,而在Al原子内部则没有出现比较大的电子密度梯度,分析Al原子内部基体成键特点可知,在金属内部会形成金属键,自由电子属于全体Al原子共有,因此在Al原子基体内部电子密度较为均匀。在界面处,C原子和Al原子以及C原子和Si原子之间均有电子云的重叠,而C原子和Si原子间重叠较为强烈,表明界面结合可能是以Si和C的结合为主要方式。通过差分电荷密度图可进一步研究原子间成键情况。 图4 Al-C端顶位界面电荷密度分布 图5所示的Al-C端顶位界面差分电荷密度图表现了界面上各原子结合时得失电子情况,其中蓝色代表失电子,白色代表不变,红色代表得电子。从图5(a)可以看出C原子在Si原子方向上形成了强力的共价键,并且由于电负性较大,共用电子对也就更偏向于C原子。电子从金属Al一侧迁移到C原子周围,显示出很强的离子键特性,表明界面处形成了Al-C离子键。第一层Al原子与第二层Al原子之间出现了一个比较强的波浪状电子聚集带,这表明在这个部分出现了比较明显的电子聚集现象。推测此时的C原子相对Al原子呈现出了正电性,而Al原子相对C原子呈现出了负电性。界面结合强度的主要来源可能是这之间的正负吸引力。 图5 Al-C端顶位界面差分电荷密度图 1) 文中基于密度泛函理论的第一性原理获得了SiCp/Al基复合材料界面结构,计算并分析了SiCp和Al各低指数面的表面能、Si和C端界面形成焓、结合强度、断裂韧性和布居数。 2) Al原子中的(111)晶面具有最低的界面表面能,具有最好的界面稳定性,在形成Al与SiCp的相界面时由Al(111)晶面与SiCp(0001)晶面形成相界面的可能性最高。Al-C端顶位界面结构界面能为0.43 J·m-2,界面强度最高。而其他的三种界面结构的界面能都在0.2 J·m-2以下,界面强度比Al-C端顶位界面的强度都要低。结合界面形成焓以及界面断裂韧性可得,Al-C顶位界面形成焓值最小,界面断裂韧性最大,即界面结构相对最稳定。因此在制备SiCp/Al基复合材料时,形成的Al-C端顶位界面会比其他三种界面更加稳定。 3) 利用差分电荷密度图分析发现,强度更高的Al-C端顶位界面上形成的结合键倾向于离子键和共价键的特性,并且在接近SiCp的Al基体中出现了强度比较高的金属键。2.3 SiCp/Al基复合材料界面强度

2.4 SiCp/Al基复合材料界面断裂韧性
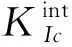
2.5 界面Mulliken布居
2.6 Al-C端顶位界面差分电荷密度分析
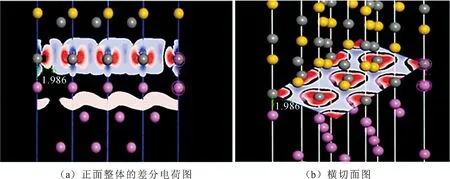
3 结 论
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
山东理工大学学报(自然科学版)(2021年6期)2021-07-02 06:31:50
铝加工(2020年6期)2020-12-20 14:08:41
西部交通科技(2020年3期)2020-06-19 08:52:47
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
中国海上油气(2019年5期)2019-10-24 03:41:08
考试周刊(2018年39期)2018-04-19 10:39:44
西南交通大学学报(2016年4期)2016-06-15 20:29:37
公路与汽运(2016年3期)2016-06-08 03:29:39
