
基于重测序的41份芝麻种质资源全基因组序列分析
2022-06-27 03:59宋成孝舒中兵邓宽平
种子 2022年5期
宋成孝, 舒中兵, 邓宽平, 金 松, 覃 成,2
(1.遵义市农业科学研究院/遵义市作物基因资源与种质创制重点实验室, 贵州 遵义 563006;2.遵义职业技术学院现代农业系, 贵州 遵义 563006)
芝麻为胡麻科胡麻属植物,栽培历史悠久。芝麻中富含各种不饱和脂肪酸、糖类、蛋白质、维生素、矿物质等,营养及药用价值较高[1-2]。芝麻原产中国云贵高原,是喜温植物[3],全球芝麻种植面积约为280万hm2,年产量约为308万t,其中亚洲占比63.2%。中国芝麻常年种植面积达70万hm2,产量约62.4万t,河南、湖北、安徽、江西、河北等省均有种植[4]。深入开展与芝麻种质相关的分子生物学研究,对芝麻分子育种及新品种的选育具有重要意义。
近年来,随机扩增多态性DNA(Random Amplified Polymorphic DNA,RAPD)、简单重复序列(Simple Sequence Repeat,SSR)、简单重复序列区间(Inter-Simple Sequence Repeat,ISSR)、扩增片段长度多态性(Amplified Fragment Length Polymorphism,AFLP)、相关序列扩增多态性(Sequence-Related Amplified Polymorphism,SRAP)等分子标记的开发应用,极大地推动了芝麻遗传多样性以及数量性状位点定位的研究[5-9]。目前芝麻全基因组测序工作已完成,与芝麻基因组相关的研究进展十分迅速。但是由于在区域性的种质资源中分子多态性标记的比例较低,遗传多样性较窄,所以应该继续开发具有高多态性的分子标记,为今后芝麻优良性状的精细定位奠定基础[9-10]。
全基因组重测序技术是针对已测得基因组序列的个体进行基因组测序,并将结果与已有基因数据库进行比对,从而发现该个体基因组内的插入、缺失突变、核苷酸序列的变化情况或者核苷酸的变异等信息,再根据结果对多种突变进行开发利用,为作物的种质筛选提供便利[11]。SNP标记是单个核苷酸变异引起基因组水平上的DNA序列的多态性, InDel是根据不同个体基因组同源序列发生的核苷酸片段插入或缺失而开发的[12-14], SV不同于前两者,是一种由大小超过50个碱基的基因组片段影响基因组序列重新排列的现象。SV包括大片段的插入、缺失、倒位、复制和拷贝数变异等[15-16]。目前,高通量测序技术使对物种基因组进行细致分析成为可能,已经广泛应用于谷子[17]、大豆[18]、马铃薯[19]等作物的遗传变异分析。
本研究以芝麻基因组为参考,对41份不同芝麻种质进行重测序,结合生物信息学分析对其进行群体遗传学分析和群体变异检测,挖掘SNP、InDel、SV等的数量及位置信息,为开发芝麻重要性状分子标记和探索种内丰富的遗传多样性奠定基础。
1 材料与方法
1.1 材 料
以来自贵州不同地区的41个芝麻地方品种为材料(详见表1)。每个地区品种随机取15粒种子,选择大小适宜的花盆,并将芝麻种子种植在花盆中,植株茎长5~6 cm时,将其茎剪下,快速封装在做好标记的离心管中,并置于超低温冰箱中保存备用。

表1 41个芝麻品种编号及来源Table 1 Numbers and sources of 41 sesame varieties
1.2 方 法
1.2.1芝麻基因组DNA的提取
首先用液氮处理41份芝麻材料,再用CTAB 法提取芝麻基因组DNA,最后用0.8%琼脂糖凝胶电泳检测DNA样品的质量[20]。
1.2.2文库构建与测序
将基因组DNA用酶随机打断成短的DNA片段后,进行平末端修复,然后在DNA片段两端连接(adapter,dA)尾,并连接测序接头。加上接头的DNA片段经过AMPure XP磁珠进行纯化,并选择300~400 bp范围的片段进行PCR(Polymerase Chain Reaction)扩增。建好的文库经过纯化、库检,Hiseq X10 PE 150上机测序。
1.2.3测序质检
测序得到的原始图像数据经碱基识别(base calling)转化为序列数据,称之为 raw data 或 raw reads。为了保证数据质量,要在信息分析前对原始数据进行质控,并且通过数据过滤来减少数据噪音。对下机的待分析数据(clean reads)再进行更严格的过滤,得到高质量的待分析数据(High quality clean reads),用于后续的信息分析。过滤的步骤为: 1) 去除dA,保留剩余的数据; 2) 去除含 N 比例大于10% 的数据; 3) 去除低质量数据 (质量值 Q≤20 的碱基数占整条 read 的 50% 以上)。
1.2.4与参考基因组进行比对统计
数据过滤后,使用微生物重测序分析软件(Burrows Wheeler Alignment,BWA)将过滤后的数据在参考基因组上进行比对,获得SAM格式的比对结果,利用samtools (用于操作sam和bam文件的工具合集) 将比对结果输出BAM格式文件,最后使用 Picard (1.129)软件标记重复reads,统计标记后的reads深度及覆盖度,再使用bedtools (v 2.25.0)软件进行覆盖度统计。
1.2.5基因组变异检测
根据比对定位结果,通过GATK软件进行包括重新局部比对和碱基质量重新校正的数据预处理,以获得高质量的可靠的SNP变异和InDel变异。此外,利用软件Manta (1.4.0)检测大片段结构变异,使用cnvnator (0.3.2)进行拷贝数变异的检测[21]。
1.2.6注释SNP、InDel和SV
使用ANNOVAR对检测出的Variant进行SNP、InDel的注释。依据变异位点在参考基因组上的位置,获得变异发生区域以及是否为同义突变等信息。
1.2.7群体分析
群体结构分析对理解进化过程提供很大的帮助,还可以通过基因型和表型的关联分析明确个体基因所属的亚群。根据以上分析得到的SNP信息,利用plink 和 frappe 软件进行群体结构分析,利用treebest软件通过邻接法构建进化树,然后基于个体间SNP差异程度使用R软件进行主成分分析。
2 结果与分析
2.1 基因重测序分析
41份芝麻材料进行全基因组重测序的数据经过滤质检后结果如表2所,共获得257.804 GB的待分析数据,经严格过滤后共有255.814 GB的高质量有效数据用于后续分析。每个芝麻品种的高质量有效数据处于5.51~7.28 GB之间,Q 20≥94.61%,Q 30≥87.12%,C/G含量处于38.98%~40.56%之间。进一步利用BWA软件将过滤后的数据与芝麻参考基因组Sesamum_indicum_v 1.0进行比对(表3),41个样品数据定位到芝麻参考基因组的数据数目占数据总数的92.69%以上,其中比对上基因组双端比对数据数目占数据总数的75.16%以上,比对到基因组上完全匹配的数据数目至少占数据总数的73.28%。结果表明,该测序结果数据信息量充分、质量良好,能够为后续数据分析提供保障。
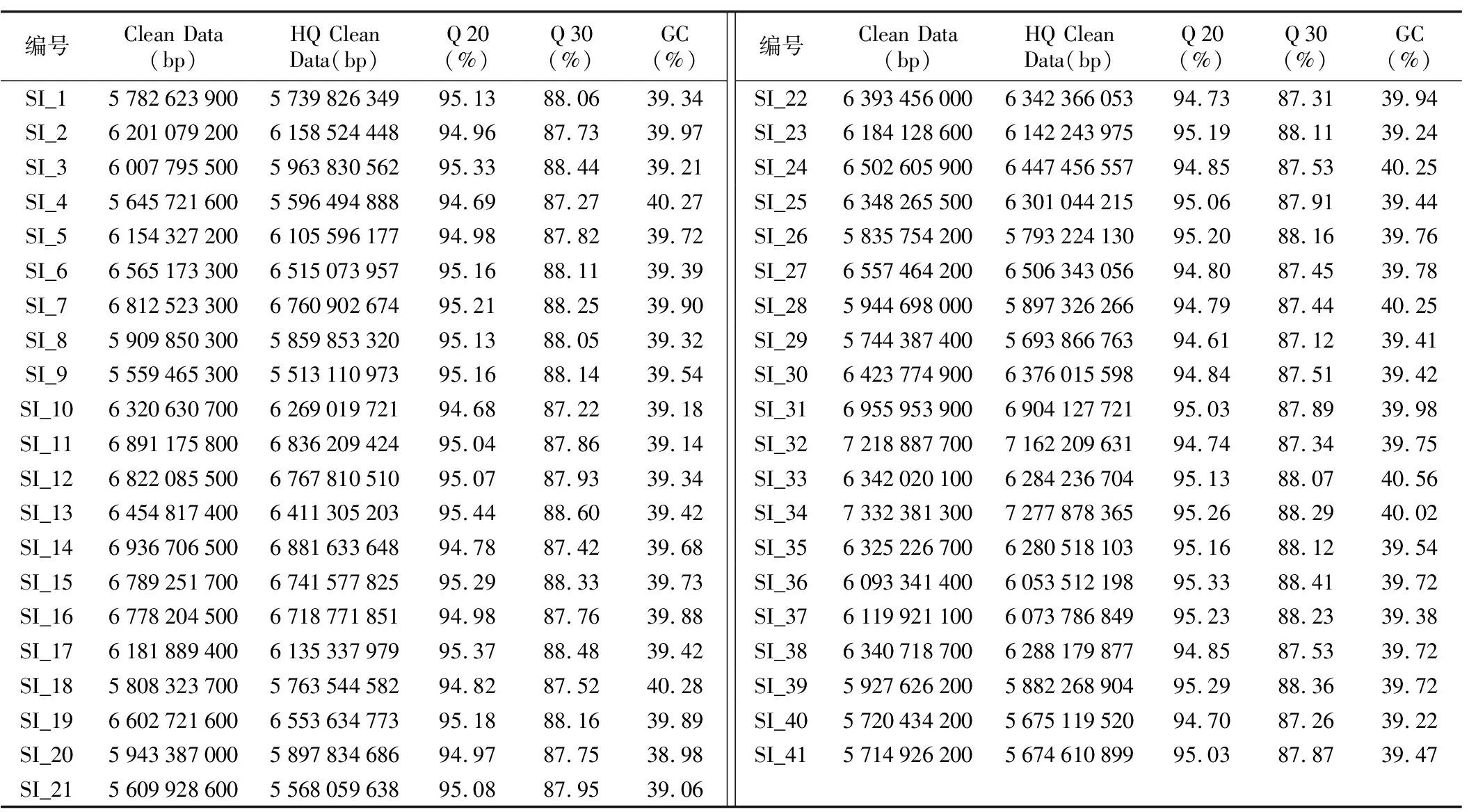
表2 测序数据的质量分析Table 2 Quality analysis of sequencing data

表3 基因组比对统计与覆盖度统计Table 3 Statistics of genome alignment and coverage
2.2 检测与注释SNP
41个样品基因组进行SNP检测并统计数据,结果见表4。其中SI_35的SNP数量最多,为1 032 613;SI_39的SNP数量最少,为215 759。41个芝麻品种中,转换型 (ts) SNP数量与颠换型 (tv) SNP数量的比值约为1.78,SI_2的ts/tv最高,SI_20的ts/tv最低;SI_8、SI_24、SI_20、SI_21、SI_29、SI_27、SI_9、SI_10和SI_39的SNP杂合比率均低于50%,分别为15.84%、17.41%、20.64%、24.14%、25.02%、25.04%、37.41%、43.12%、46.99%;SI_30、SI_15、SI_33、SI_22、SI_34、SI_23、SI_38、SI_31、SI_41和SI_40的SNP杂合比率均高于70%,分别为72.49%、72.64%、73.57%、73.58%、80.15%、82.56%、83.09%、84.74%、85.48%、86.10%,杂合比率越高,说明样品的杂合程度越高。通过ANNOVAR软件分析芝麻全基因组SNP的注释,获得变异位点在基因组发生的位置或变异类型(图1)。编码区内发生多肽产物的氨基酸序列改变或功能性RNA碱基序列改变的 SNP,易导致基因功能的变化,从而出现形态和性状的变化,41份芝麻品种发生在外显子区域的SNP总数占总SNP数量的7%,其中非同义突变与同义突变分别占48%和51%。其中,SI_31发生在外显子区域的SNP总数为77 154,在41个品种中最多,非同义突变与同义突变分别占46.83%和52.23%,且非同义突变的SNP数量为41个芝麻品种中最多;SI_39发生在外显子区域的SNP总数为14 953,在41个品种中最少,非同义突变与同义突变分别占50.82%和47.98%,且非同义突变的SNP数量为41个芝麻品种中最少。非同义突变的占比高低与品种的性状变化程度应该是一致的。

表4 41个芝麻品种SNP数据统计Table 4 SNP data statistics of 41 sesame varieties

图1 41份芝麻SNP位置和功能注释Fig.1 SNP location and functional annotation of 41 sesame varieties
2.3 检测和注释InDel
在全基因组范围检测过程中,发现芝麻的41个品种检测出的InDel总数范围为31 819~144 363(表5),其中SI_35的InDel总数最多,SI_39的InDel总数最少,InDel数量的变化规律与SNP基本一致。SI_31、SI_40、SI_41、SI_23、SI_38、SI_34的InDel杂合比率均高于70%,分别为78.70%、76.24%、75.96%、75.91%、72.11%、70.90%;SI_29、SI_27、SI_21、SI_20、SI_24、SI_8的InDel杂合比率均低于20%,分别为18.85%、18.84%、16.21%、13.44%、11.41%、10.31%。通过与芝麻基因组进行比对,注释出41份芝麻InDel位点发生的位置以及其突变的类型(图1)。有54%的InDel发生在基因间区,20%的InDel发生在内含子区域,2%的InDel发生在外显子区域。其中,SI_35发生在外显子区域的InDel数量最多,为2 689;其次是SI_31,为2 643;SI_39发生在外显子区域的InDel数量最少,为1 115。InDel在外显子区域的突变包括移码突变和非遗码突变,41个品种发生移码突变的比例在54.85%~59.82%之间。
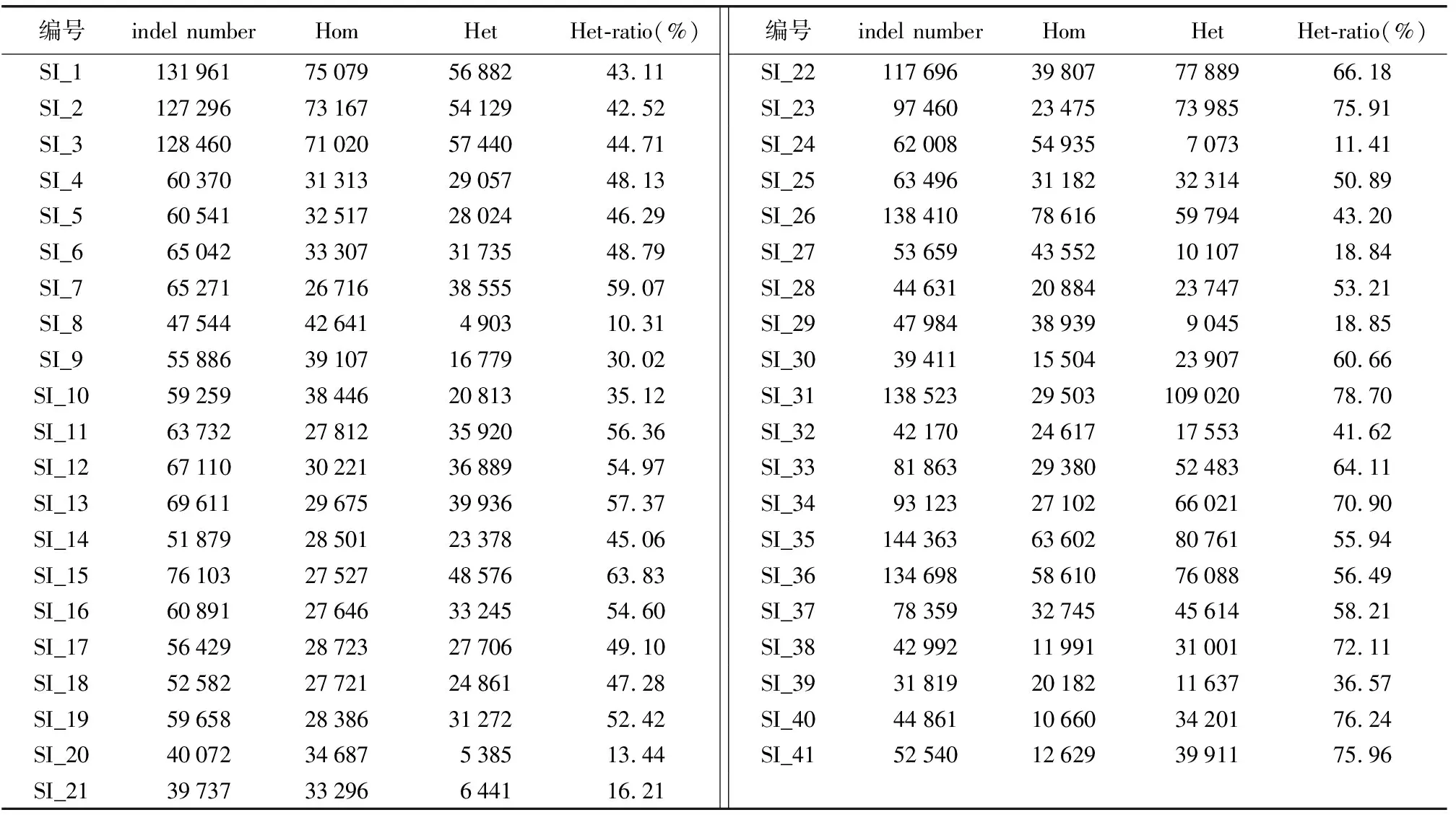
表5 41个芝麻品种InDel数据统计Table 5 InDel data statistics of 41 sesame varieties
2.4 检测和注释SV
利用Manta (1.4.0)软件检测SV与参考基因组进行位置信息比对并注释其变异类型,共检测得到41个样品各类型SV共计138 135个(表6),SI_35的SV位点数量最多,为5 328个,其中插入(insert)和缺失(delete)变异占总变异的42.81%,染色体间异位(interchromosomal translocation)变异占总变异的44.97%,串联重复(Tandemrepeats)和倒位(inversion)变异占总变异的12.22%;SI_41的SV位点数量最少,为2 395个,其中插入(insert)和缺失(delete)变异占总变异的28.14%,染色体间异位(interchromosomal translocation)变异占总变异的55.70%,串联重复(Tandemrepeats)和倒位(inversion)变异占总变异的16.16%。在结构变异的5种类型中,染色体间异位(interchromosomal translocation)类型的变异位点数量远超过另外的4种类型,约占每个芝麻品种SV位点数量的1/2;其次是缺失类型,而串联重复类型最少。缺失类型的变异位点数量约为插入类型的2倍。41个芝麻品种的SV位点的数量变化规律与SNP和InDel不一致。
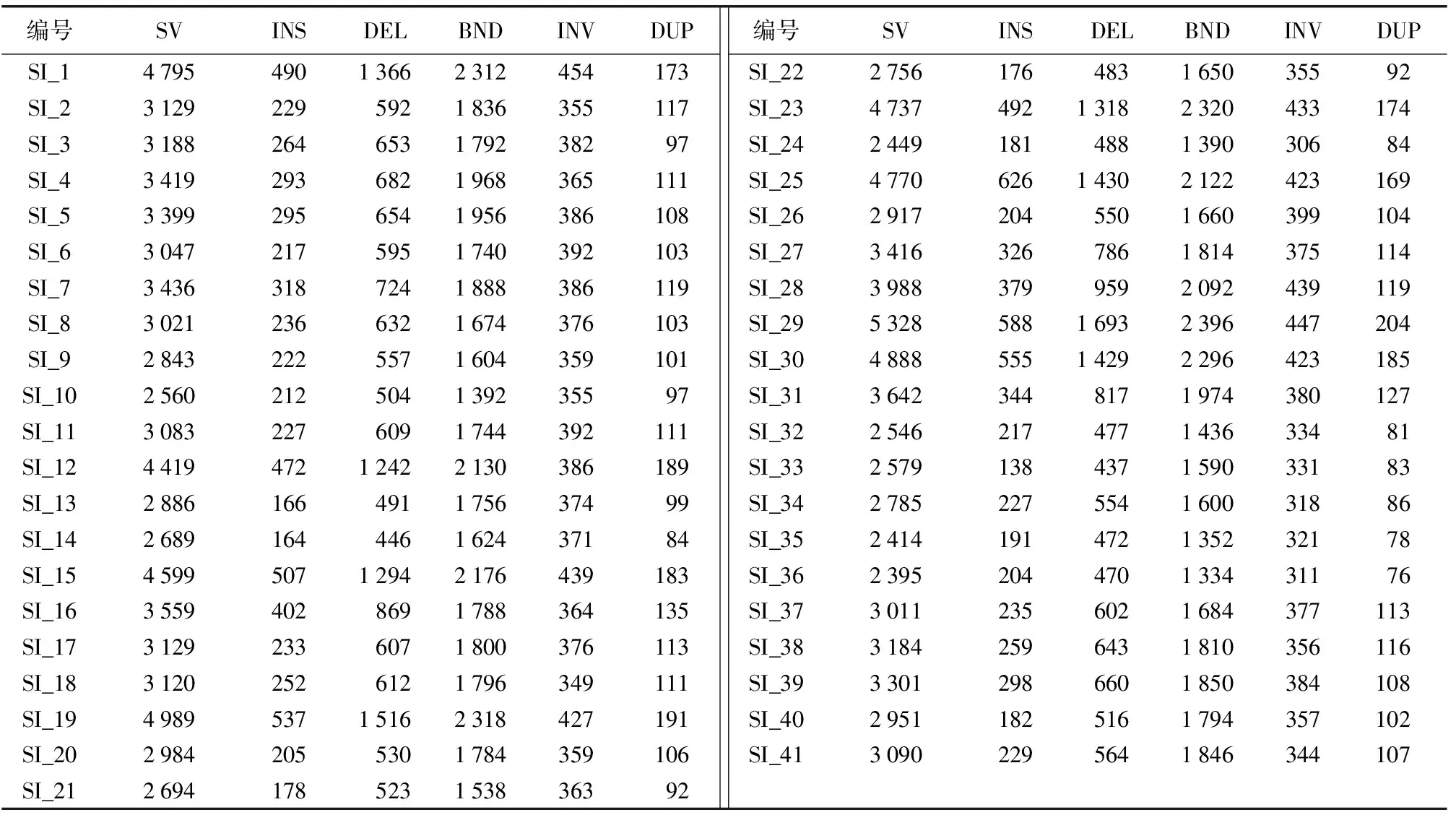
表6 41个芝麻品种SV数据统计Table 6 SV data statistics of 41 sesame varieties
2.5 芝麻种群结构分析
2.5.1芝麻种群遗传结构分析
基于以上分析得到的SNP数据信息,利用plink和frappe软件进行群体结构分析,并对不同K值的交叉验证错误率进行比较,结果见图2。当K=4时的错误率均较低,说明模型的可靠性较好。另外对不同K值的模拟分析结果如图2 A:K=2时可见41个芝麻品种种群最深的分化为两个谱系种群(红色和蓝色);K=3时两个谱系种群开始进一步分化,此时约有20%的品种含有3个谱系的遗传成分;K=4时约有36.6%的品种含有4个谱系的遗传成分。总体来看,遗传成分相似且较稳定的品种分为4组,41份芝麻种群至少有4个祖先遗传成分。

图2 不同K值模拟的交叉验证错误率(A)与芝麻种群遗传结构(B)Fig.2 Cross validation error rate (A) and genetic structure of sesame population (B) simulated by different K values
2.5.2芝麻主成分分析
采用主成分分析方法研究41个样本间的遗传背景相似性和聚类关系,其中PC 1和PC 2、PC 3的贡献率分别为25.79%和13.61%、8.93%,PC 1-PC 2、PC 2-PC 3散点图如图3。由图3可知,PCA的分析结果与以上遗传结构分析结果相符,3个PCA均将41个芝麻品种分为4个谱系。

图3 41个芝麻品种PCA分析Fig.3 PCA analysis of 41 sesame varieties
2.5.3芝麻系统发育进化树构建
根据得到的SNP数据信息,计算样品间距离,对41个芝麻品种进行聚类分析得到样品进化树(如图4),其结果与主成分分析和遗传结构分析的结果相似。SI_1、SI_3和SI_36为一个谱系,SI_2、SI_22为一个谱系,SI_26、SI_35、SI_31、SI_23为一个谱系,其余SI_9、SI_10、SI_27、SI_34等32个品种为一个谱系。
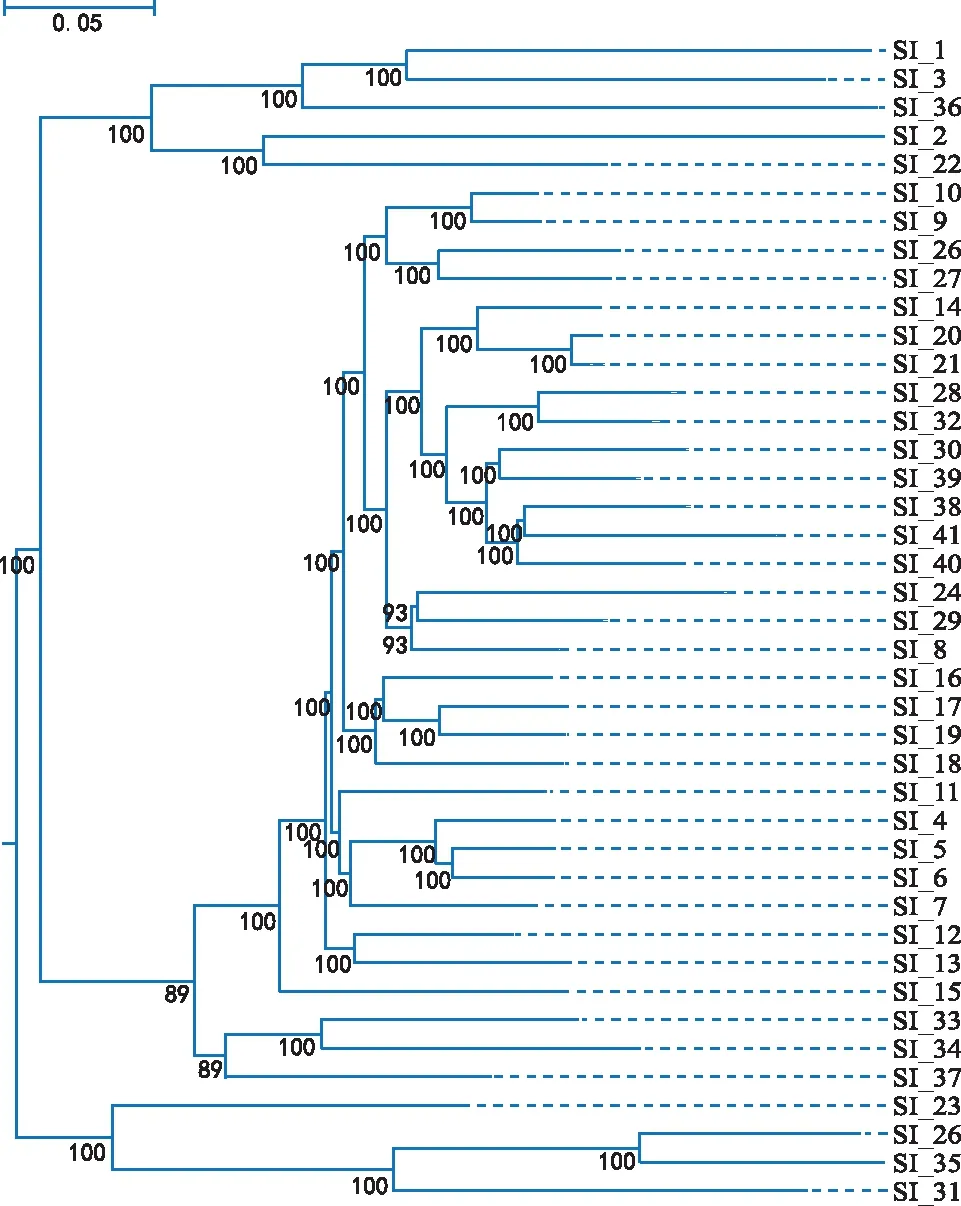
图4 41个芝麻品种系统发育进化树Fig.4 Phylogenetic evolutionary tree of 41 sesame varieties
3 讨 论
芝麻是我国主要的油料作物之一,年总产量居世界首位,其分布范围十分广泛,芝麻籽含有丰富的不饱和脂肪酸和蛋白质,营养价值较高[22]。芝麻在贵州的种植历史悠久,长期选择的结果逐渐形成了适应贵州气候环境的芝麻种质资源,但不同生态区的芝麻种质资源之间性状差异较大,具有丰富的遗传多样性,故贵州芝麻种质资源有很大的利用空间和筛选潜力[1]。
目前,针对芝麻遗传多样性、生理学特性、分子标记开发、遗传图谱建立、重要的性状基因挖掘与精准定位、全基因组和转录组基因的测序等方面已取得很大的突破性进展。收集贵州产地41个代表性芝麻品种资源,观察主要形态、性状,并对其遗传多样性和性状间的相关分析进行评价,为贵州产地的芝麻品种的分类、资源的利用以及遗传改良提供参考。
Nava,Gerder[23]利用电泳对委内瑞拉5个栽培品种的种子以及幼苗叶片的蛋白质提取液进行分析后发现,酯酶、酸性磷酸酯酶、过氧化物酶、苹果酸脱氢酶等的电泳图谱有表现型的变异存在,赵应忠,周明德[24]研究表明,脂酶在品种个体之内存在多样性差异,这个结果对芝麻生态区的划分有一定的参考价值。利用连锁分析发现,芝麻种皮颜色是被2个加性显性主效基因和上位性多效基因控制,并成功定位到4个相关的数量性状基因位点(quantitative trait locus,QTL)[25],而且芝麻株高相关性状的遗传复杂性定位到26个与株高相关的QTL位点[26]。关联分析在芝麻性状研究上应用较广泛,Wei等[27]选取216份芝麻核心种质,用于研究芝麻中种子的蛋白质、亚油酸等含量的相关性,从中发现了与种子含油量显著相关的8个分子标记。随着测序技术的快速发展,全基因组重测序和转录组在芝麻基因组以及分子标记开发等研究上也得到广泛的应用,Wei等[28]对705份国内外材料进行基因组重测序,开发出SNP标记达500多万个。另外利用转录组对不同芝麻品种的现蕾期和种子发育的过程进行研究,得到与芝麻生长发育和种子形成密切相关的基因序列。
本研究通过对41个芝麻种质资源进行重测序并与参考基因组进行比较,初步统计了每个品种在全基因组范围内发生的变异类型及数量,对SNP、InDel、SV进行检测和注释,并根据得到的SNP信息对41个种质资源进行种群结构分析。SNP的变异类型分为转换(ts)和颠换(tv),各芝麻品种的ts/tv比例均在1.78左右,表明转换类型的变异比颠换更容易发生。杂合SNP位点数量占各品种总SNP数量的比率没有一致性,说明41个芝麻种质资源中杂合程度有高有低。从InDel位点数的结果可以看出,各品种芝麻样品与芝麻参考基因组之间存在差异,编码区的InDel会引起移码突变,从而导致基因发生功能上的改变。SV有缺失(delete)、插入(insert)、倒位(inversion)、串联重复(Tandemrepeats)、染色间异位断点(interchromosomal translocation) 5种类型的结构变异,其数量反映出基因组序列大片段的缺失、插入、导致等变异程度。种群结构分析可以表出41份芝麻种质之间亲缘关系的远近程度以及种群进化过程。本研究发现的这些分子标记可为芝麻的遗传多样性分析、优良性状基因定位以及优良品种选育提供基础。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
中国种业(2022年1期)2022-01-27
中国种业(2022年1期)2022-01-27
童话王国·文学大师班(2020年8期)2020-09-27
作文与考试·初中版(2020年18期)2020-06-19
电脑知识与技术(2018年19期)2018-11-01
