
基于量子化学计算柴油在CO2/O2氛围下的燃烧特性
2022-06-24 07:57王龙刘永峰毕贵军宋金瓯
化工进展 2022年6期
王龙,刘永峰,毕贵军,宋金瓯
(1 北京建筑大学,北京市建筑安全监测工程技术研究中心,北京 102616;2 新加坡科技研究局制造技术研究院,新加坡 637662;3 天津大学内燃机燃烧学国家重点实验室,天津 300072)
近年来,温室效应不断累积,导致地球表面温度上升,造成全球气候变暖。1992 年联合国制定《联合国气候变化框架公约》来应对这一环境问题,随后通过《京都议定书》对《联合国气候变化框架公约》进行具体化,其中减少温室气体CO的排放更是应对气候变暖的重要措施。并且因为氮氧化物会积极参与臭氧消耗、光化学烟雾和酸雨的形成,所以减少氮氧化物、碳氧化物和未燃烧的碳氢化合物的形成是一项具有挑战性的任务。液氧固碳闭式循环柴油机实现了“零排放”,其主要是将柴油机尾气中一部分CO固化成干冰,而未固化的CO重新进入进气系统成为燃烧背景气体的一部分。因此就形成了CO/O的燃烧背景,而目前针对CO/O氛围燃烧的研究大多数是煤粉的燃烧,或是小分子物质的燃烧。对于柴油在CO/O氛围下的燃烧鲜有报道,CO/O氛围下的燃烧特性和机理有很大的研究空间,本文则对柴油在CO/O下的燃烧特性进行研究报道。
相关科研工作者对柴油燃烧做了大量的研究,而火焰浮起长度在燃烧与碳烟形成过程中有显著影响,因为它与燃烧区域上游的燃料-空气混合过程密切相关,也与喷雾过程和着火延迟有很大关系。Siebers 等从实验角度对柴油燃烧火焰浮起长度进行考察,提出一种OH化学发光(波长310nm)成像技术来确定火焰浮起长度,从环境气体密度(7.3~58.5kg/m)、环境气体温度(800~1300K)和氧气浓度(15%~21%)来考察环境对火焰浮起长度的影响。结果表明,环境气体温度、密度或是氧气浓度的升高,都会引起火焰浮起长度变短,因此Siebers等给出了空气卷吸率。不仅如此,他们还研究了氧气浓度引起的浮起长度变化对火焰上游燃料-环境气体混合量和碳烟形成的影响,结果表明,浮起长度和氧气浓度成反比,而先前的观察并没有得到这种反比结论,这一趋势表明燃料燃烧和蒸发过程之间的相互作用将会减少。随后,Siebers等提出了“氧比”的概念,将浮起长度引入空间积分自然光度(spatially integrated natural luminosity,SINL),将SINL作为燃料类型和浮起长度总氧比的函数,从而进一步探究含氧量对浮起长度的影响以及和碳烟生长的关系。结果表明浮起长度上游发生更多的空气输送和空气混合,碳烟水平随着氧比的增加而降低。Siebers 等在火焰浮起长度上做出了突出的贡献。Eismark等在火焰-壁面相互作用的高压高温燃烧室中进行不同柴油燃烧的光学研究,结果表明不同燃料的火焰浮起长度都相似,但低碳烟的浮起长度都比较大,而氧气增加了喷嘴附近的内部稀释,减少了碳烟的形成。Pastor等同时采用三种成像技术(高速摄影直拍自然光度、纹影法,OH化学发光)进行燃烧特性实验,实验表明火焰浮起长度随环境温度和密度的升高而变短,这是因为减少了燃料燃烧所需的空气量。Eagle 等基于Siebers 所做的定容燃烧弹实验,设计了新的发动机实验,其具有三个90μm的喷孔和光学通道。测量的液体长度、着火延迟和火焰浮起长度与之前Siebers 的实验数据进行比较,并没有出现无法解释的异常值,结果趋势高度一致,这表明液体长度浮起的瞬态行为主要是由于内部喷射器动力学。Yin 等研究柴油在空气氛围和预混合甲醇-空气混合氛围中的燃烧特性,他们表示所有实验下,火焰浮起长度随环境温度的升高而降低,一旦形成主反应区,火焰就会同时往下游和喷嘴传播,火焰浮起长度首先降低,然后达到准稳态,而甲醇对着火延迟和火焰浮起长度都有抑制作用。Kahila 等利用大涡模拟方法,对正十二烷进行宽范围压力实验,结果表明火焰浮起长度随注入压力的增加而增加,着火延迟随注入压力增加而减小,特别是他们引入准稳态扩散火焰中的FGM trajectory分析来解释火焰浮起长度实验和数值计算之间的误差原因非常有趣。火焰浮起长度的影响因素主要分为环境气体密度、喷射压力、环境温度以及氧浓度等。一些研究人员进行了更广泛的研究,Tagliante 等使用激光诱导等离子体在不同环境温度(800~850K)和喷油轨压(100~150MPa)下进行点火,利用光学诊断技术来研究单孔喷嘴喷射柴油的燃烧情况,采用纹影法和OH化学发光法来跟踪甲醛位置和火焰浮起位置。结果表明,温度越高,火焰浮起长度越短,浮起长度下游主要是低温反应的主导。为了更好地研究火焰燃烧稳定机制,Tagliante 等采用定容燃烧弹实验和直接数值模拟(DNS)的方法,再现柴油机中燃料到空气中产生的扩散火焰现象,得到了火焰拓扑结构和火焰浮起长度,表明了火焰浮起长度会迅速跳到较小值,然后产生的火焰传播到下游。
以上对柴油在空气氛围下的燃烧特性都做了深入的研究,但本文作者发现很少甚至没有柴油在CO/O氛围下燃烧特性的研究。因此本工作提供一项柴油在CO/O氛围下燃烧的基础研究,尤其是高浓度CO对柴油表征燃料燃烧的影响。通过光学定容燃烧弹实验台架和高速摄影机对柴油火焰传播进行记录,利用量子化学计算完善现有机理,利用CFD软件进行数值仿真,将实验结果与计算结果进行对比分析,得到有趣新颖的结论。
1 机理模型建立
1.1 CO2高温裂解及产物(CO)生成OH自由基
采用电子相关的耦合簇理论方法,其包括一个给定类型的相关到无穷阶,相当于多体微扰的全部项,考虑对一个参考态的波函数加上所有类型的相关到给定阶数。特点是将高激发组态的展开系数和低激发组态的展开系数耦合,然后通过迭代求解一组非线性方程组来得到组态波函数的展开系数以及总能量。采用CCSD(T)/aug-ccpVTZ对CO—→— CO+O·进行柔性扫描。将碳-氧原子在5.35Å(1Å=0.1nm)处完全断裂产生对称破缺波函数,CO 分子和O 自由基对应的能量为-493438.80kJ/mol,碳-氧距离逐渐减小,之后每一步都会采用上一步所得到的收敛波函数作为初猜,当距离缩小到小于“不稳定点”后,此时基态波函数就是闭壳层波函数。而CO高温裂解出的CO 可以反应生成OH 自由基,即O+CO+H·—→— CO+OH·,如图1 所示。而CO裂解计算采用的是电子相关方法中的耦合簇理论结合相关一致基组来获得高精度的能量,这种方法只限于在一些小分子体系中使用,并且没有解析梯度不能用于过渡态的计算。从头算理论是从薛定谔方程的波函数出发,对原子和分子体系进行了近似描写,但是波函数并不是可观测量,只有电子密度是可观测的,其体系的基态能量由密度唯一确定,所以用电子密度来描写体系,即密度泛函理论(DFT),此过程采用B2PLYP-D3(BJ)/def2tzvp。如 图1 路 径1 所 示,O与H自由基都会结合在CO分子的C端,形成中间体1,比反应物的自由能高出279.21kJ/mol,而氧端具有结合H自由基的能力,因此结合在碳端上的H、O 有着断键成键的趋势,经过渡态1(相对能量为352.64kJ/mol),再经过IRC路径分析和产物结构优化生成中间体2(CO和OOH),此时CO的碳端仍具有成键能力,可以与OOH 的O形成C—O,因此可以将C 与O 拉近搜索下一个过渡态结构。经过计算,过渡态2存在唯一虚频,且虚频振动方向与预测的成键方向完全一致,为正确的过渡态结构,Δ为340.08kJ/mol。再对过渡态2 进行IRC 路径计算和产物结构优化,形成中间体3,Δ为282.58kJ/mol,此结构已十分接近目标产物CO和OH 自由基,但O—C—O 仍然存在夹角,并不是CO分子的直线结构,经过观察与测量,此时类似CO的C 与O 距离在1.6Å 附近,在量子化学中,此距离表示存在相互作用,有成键趋势,因此将此距离拉近1/3进行过渡态搜索,对初猜过渡态进行优化,则形成过渡态3结构,Δ为310.41kJ/mol。对过渡态3 结构进行IRC 路径分析,产物为CO分子直线结构和OH自由基,再对此进行结构优化得到最终产物。如图1 路径2 所示,O与CO 的C 端结合,H 自由基与CO 的氧端结合,形成中间体1,Δ为472.14kJ/mol。要形成OH 自由基,就要有H自由基与O中的O结合的趋势,因此将H自由基拉到O附近作过渡态搜索,发现唯一虚频,对过渡态结构进行优化得到过渡态1,Δ为550.41kJ/mol。对过渡态1 进行IRC 路径分析,并对产物进行优化,得到中间体2,Δ为178.10kJ/mol,此时的中间体到目标产物,需要中间体2 结构中的CO 得到氧形成CO,OOH 失去氧形成OH 自由基,因此将OOH的氧端向CO的碳端靠近,对结构进行优化得到过渡态2,对此结构进行IRC 路径分析和产物优化,得到目标产物CO和OH自由基。

图1 OH自由基生成的两条新反应路径
1.2 OH自由基与柴油高温裂解产物反应
甲苯、正庚烷分子量较大,依然采用密度泛函方法计算。如图2反应1所示,OH自由基攻击甲苯的C脱氢生成水,释放346.12kJ/mol的能量,形成苄基,然后苄基会结合OH自由基形成苯甲醇,释放2233.23kJ/mol 的能量,如图2 反应2 所示。图2反应3为OH自由基攻击苯环的C,OH自由基直接与此位点结合,通过过渡态能垒1831.72kJ/mol,形成产物CHOHCH,因为OH 自由基与甲苯苯环邻、间、对位点反应的量子化学计算,Truhlar等已经给出,本工作计算了未研究到的位点反应。OH 自由基与甲烷反应时首先进行夺氢反应,生成甲基自由基和水,如图2 反应4 所示。而OH 自由基会继续与甲基反应直接结合生成甲醇,因为大部分自由基与自由基的结合没有过渡态直接生成新的化学键。当OH 自由基与乙烯反应时,OH 自由基进攻CH上的C或C时,OH自由基会裂解成氧自由基和氧自由基形成环氧乙烷中间体,然后经过图2 反应5 的过渡态形成乙醇。当OH 自由基进攻丙烯的C或C时,也是先形成环氧丙烷中间体,然后经过图2 反应6 的过渡态,形成产物CHCHOHCH。因为C和C都是不饱和双键上的碳原子,因此无论OH自由基进攻C或C,经过计算反应路径都一样。而当OH自由基进攻C时,此处的碳—氢键为饱和键,经过计算并没有中间体,直接通过图2反应7的过渡态能垒形成丙醇和氢自由基。

图2 OH自由基与柴油热解产物反应
1.3 敏感性分析及机理简化
将密度泛函量化计算的新反应耦合到Shermo和KiSThelP,通过过渡态理论原理,假定反应物由于与环境的能量快速交换而处于热力学平衡状态,其中分布于过渡态(连接反应物和IRC产物上能量最大点)的反应物就会转化成产物,而量子化学中的隧道效应会增大反应速率常数,通常的理论计算频率是谐振频率,一般会忽视非谐振效应,因此直接基于此计算的基频、零点能、焓、熵等可能会有较大误差,因此要考虑隧道效应校正。采用劳伦斯利弗莫尔国家实验室所提出的正庚烷详细化学反应动力学(654 个组分,2827 步反应)和甲苯详细化学反应动力学(530 个组分,2808 步反应),并对其进行敏感性分析,进而考察给定条件(温度、压力和浓度)的影响程度。一个由种组分和个反应组成的系统,第个物质的反应速率可用微分方程表示为式(1)、式(2)。
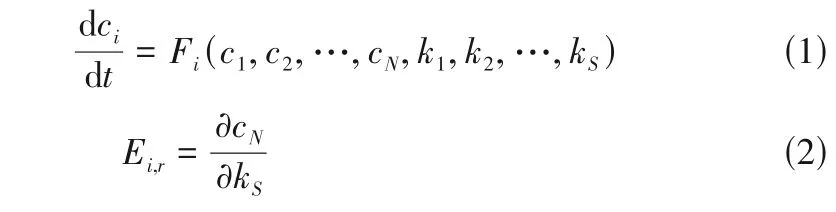
式中,c为物质的浓度;c的解对参数k的依赖程度表示为敏感性系数;k为系统参数。这样即可以通过敏感性系数来判断每个基元反应对该参数的影响程度,这是对机理简化的重要参考。采用定容绝热模型,全混反应器来模拟内燃机中柴油着火燃烧的历程,边界条件设定初始温度850K,初始压力3MPa,当量比1.0 正庚烷-甲苯在前期缓慢减少,由于O+CO+H·—→— CO+OH·的作用,着火促进剂OH自由基逐渐累积,温度缓慢增加,当达到着火点前期时,正庚烷-甲苯迅速氧化,温度剧烈增加。为避免Lu 等提出的直接关系图法(DRG)中每一个被选择的组分都具有同等的重要性和它所属的强耦合组分的集合必须完全保持的缺点,采用消除误差影响的DRGEP 方法,见式(3)~式(6)。

式中,为A对B的依赖(删除组分B对组分A的产生和消耗所造成的影响);为第个基元反应中组分A 的净反应计量系数;ω为第个基元反应的净反应速率和为直接相互作用系数。
2 实验
如图3 所示,定容燃烧弹系统主要包括燃烧室、高压共轨系统、温度控制系统、冷却系统、压力检测系统和高速摄影系统。燃烧室为圆柱形结构,外部半径265mm、外部高度810mm、内部半径150mm、内部高度560mm。燃烧室内设有喷射器、温度和压力传感器、加热瓦、进气口和排气口。燃烧室有四个光学窗口,由130mm×32mm 的高透光石英玻璃装载密封,外壁配有冷却管来冷却系统。可视化区域半径为50mm的圆形区域,能够充分观察柴油的喷雾和整个燃烧过程。实验开始时,首先进行排气,将之前实验的剩余气体或加载石英时进入的空气去除,避免无关气体影响实验。通过流量阀来控制进气速度和进气量(精度为0.01MPa)。然后对燃烧室进行预加热,模拟真实发动机内部的燃烧温度(298~850K)。设定目标温度直接加热,无法稳定控制,会对系统造成破坏,因此需要梯度加热。加热梯度刚开始为100K,当达到600K,换为小梯度50K 进行加热(加热瓦总电阻4.5Ω,最大输出功率11.0kW)。加热开始时,打开外壁冷却管,对外部组件进行冷却,预热燃烧室大约需要20min。与此同时,同步建立喷油压力,高压共轨系统由供油系统和燃油喷射系统组成,由变频电机驱动建立高压燃油泵(最大油压175MPa),采用ECU的高压共轨系统,可灵活改变喷射压力、喷射脉冲宽度、喷射方式(单、双)和喷射频率。然而,仍然需要建立梯度压力来保证实验设施的安全稳定性。油轨压力100MPa 之前梯度压力为10MPa,100MPa后的压力梯度为5MPa,可以精确控制压力,直到建立120MPa 的喷油压力。当燃烧室温度和轨压达到预设值时,调整高速摄影系统。高速摄影系统主要包括高速摄影机、主机、监视器、多通道数据集电极和信号延迟器。在石英玻璃窗的中心放置一个光源为高浓度CO氛围下火焰燃烧提供一个明亮的拍摄视野。当燃油喷射被打开,高速摄影机完全记录柴油在燃烧室中燃烧过程并将记录的数据信息发送回控制计算机和进行后处理。实验条件参数如表1所示。
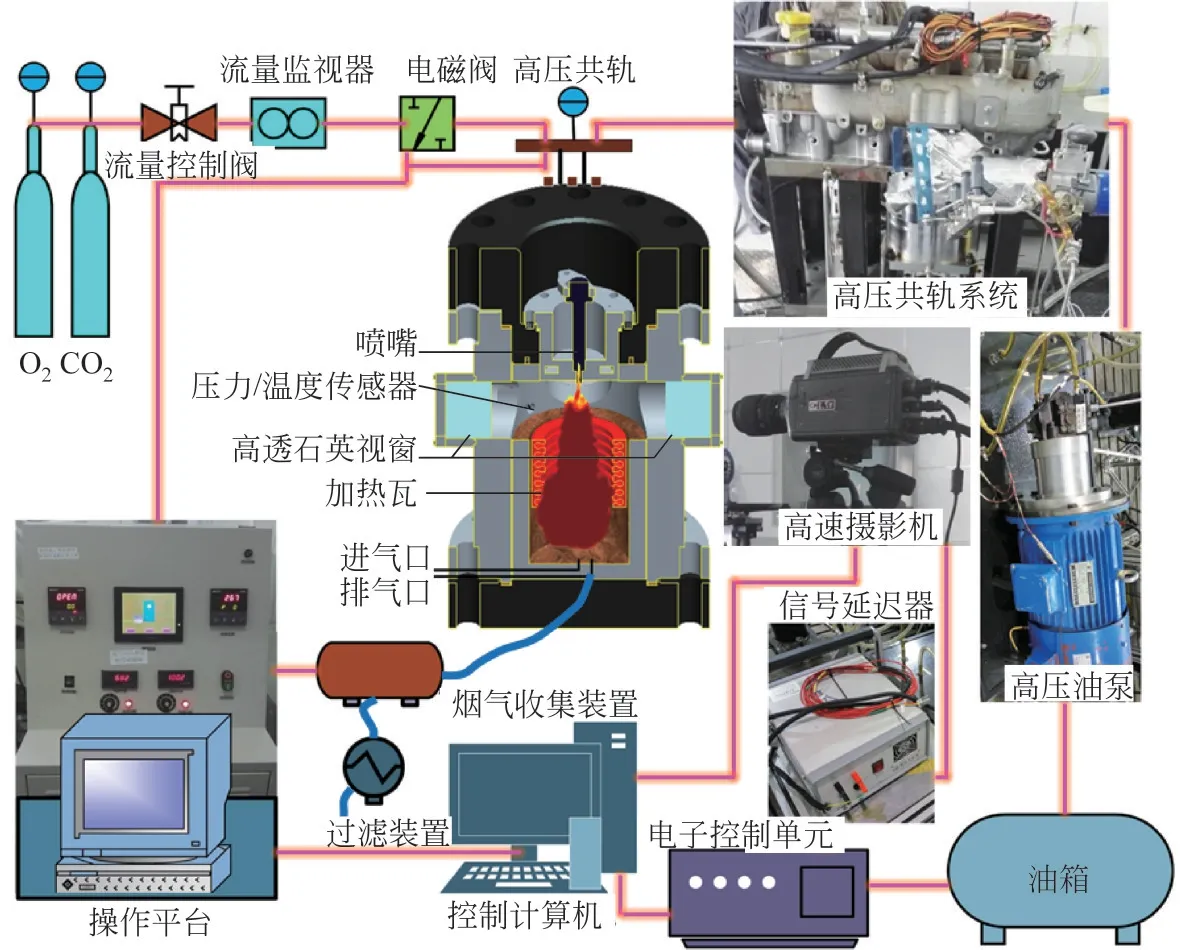
图3 实验系统
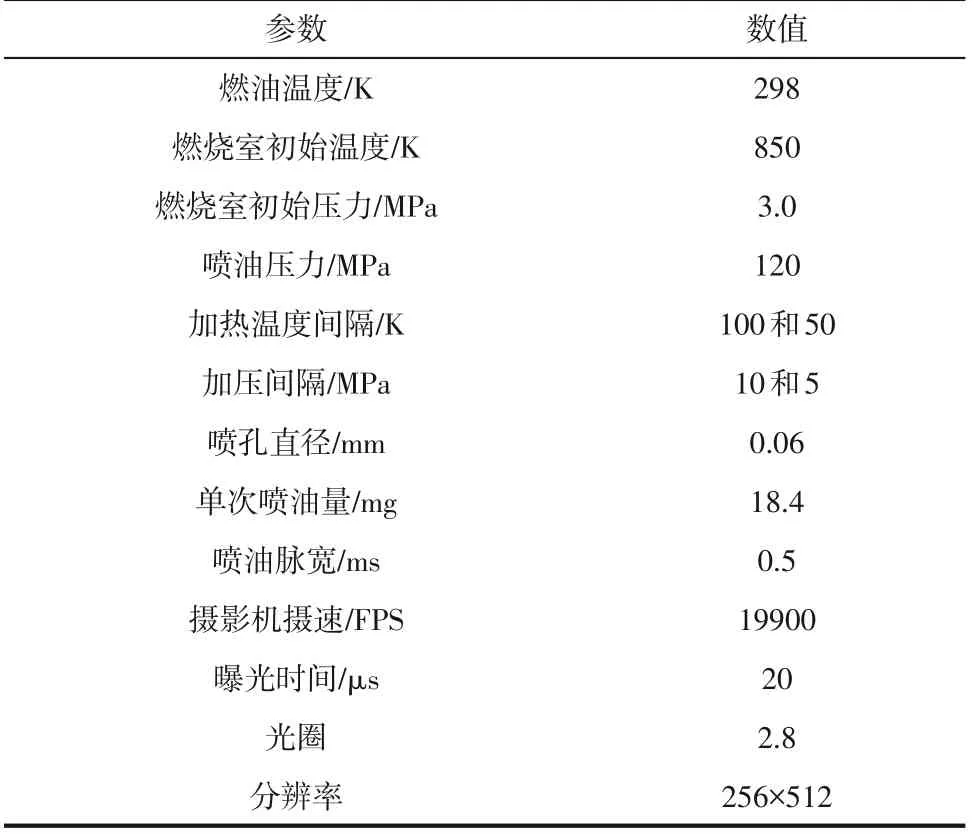
表1 实验参数设置
3 结果与讨论
3.1 一氧化碳反应位点预测
图4(a) 是一氧化碳平均局部离子化能(ALIE),图中红色区域是反应不容易发生的区域(ALIE较大),而蓝色区域则是反应较容易发生区域(ALIE 较小),也是CO 的碳端位置(ALIE为12.61eV),是整个分子的ALIE 最小的位置,即反应亲电位置。表明碳端的电子被束缚得很弱,因此碳端的活性就较高,是容易参与亲电或自由基反应的活性位点。说明氢自由基与CO 的碳原子结合比O原子结合更容易发生,证明了所提出的路径1比路径2 容易发生。虽然通过ALIE 极小点表明了CO 的碳端比氧端活性更高,但是并没有得到定量的氧端ALIE 数值,仍然具有缺陷,因此,本文把ALIE和分子表面静电势(ESP)结合,进行联合分析,得到更为科学合理的解释。通过图4(b)可以看出,红色区域是ESP为负的区域,蓝色部分是ESP为正的区域。ESP为负,说明此处静电势由电子电荷所主导,ESP为正,则说明由核电荷所主导。而CO 分子表面的ESP 负值区域在碳端和氧端,而在碳原子和氧原子结合处的ESP 明显为正值,并且ESP极大值为0.54eV,由于考虑到反应活性,所以ESP极大值点并不是本工作所感兴趣的点。而ESP极小值点在CO 分子的碳端(-0.51eV),说明此处的活性最大,这与ALIE 分析的结论相一致。虽然CO 分子氧端ESP(-0.37eV)比碳端稍大一点,但仍然为负值,说明了在氧端依然具有活性,这解释了在计算氢自由基进攻CO分子时,会同时进攻CO分子的碳氧两端。而碳端的ESP最小,相对于氧端更能吸引电子,这也解释了氢自由基会优先结合CO分子碳原子端。虽然H自由基结合CO的碳氧原子能力有优先顺序,但在高温高浓度下运动和碰撞会更为剧烈,所以路径1和路径2都会发生,并且最后形成的二氧化碳和着火促进剂OH自由基的相对自由能变为负,从反应物到产物能量降低,也符合化学反应动力学。

图4 一氧化碳平均局部离子化能和表面静电势
3.2 OH自由基与柴油热解产物反应位点分析
图5 为甲苯的ALIE 和ESP,甲苯ALIE 值最小的区域分布在碳环处,其中邻位与对位出现了极值点(9.32eV),而在甲苯的甲基处也有ALIE值较小的区域,由此可以得到甲苯的碳环和甲基处的碳位是活性位点。从图5(b)看出,ESP 为负的区域主要分布在碳环处,最小值为-0.79eV。所以容易找到的甲苯的活性位点是碳环处的邻、间、对三个位置,过渡态的结构容易猜测,因此Truhlar等只给出了OH自由基在甲苯苯环处的邻、间、对三个位置的反应动力学研究,但是没有通过量子化学的定性与定量分析,且没有给出其他位置的反应研究。通过图5(a)、(b)看出,在甲苯的甲基处的碳存在ALIE 值较小和ESP 较小的位置,在甲苯碳环和甲基相连处的碳也明显有蓝色区域位置,因此推测这两处可能具有活性,可以发生反应,通过过渡态的计算,证明了这两处可以发生反应,并且在图2中给出了具体的反应路径。

图5 甲苯平均局部离子化能和表面静电势
OH 自由基与柴油热解产物(甲烷、乙烯、丙烯)反应,与甲烷反应是先脱氢形成水与甲基,与乙烯反应会先形成一种环氧烷烃结构的中间体,并不会直接形成过渡态。如图6 所示,ALIE 值较小的区域在双碳位置,极小点(9.17eV)出现在双键中间位置,ESP 为负(-0.66eV)的地方也在双键位置,因此可以判断反应会发生在双键中心,结合原子时不倾向任何原子,会形成一个正的环氧结构,即环氧乙烷,图2反应5的计算也很好地符合了这一分析。而形成的环氧乙烷ALIE 极小值(11.76eV)出现在氧原子处,氧原子处的ESP最小(-1.46eV),而在双键位置也有极值点(0.18eV),相对于其他的极值点(0.73eV)较小,但仍然是正值,不具有亲电能力,反应不容易发生。因此后续反应会与环氧乙烷的氧发生反应。

图6 乙烯平均局部离子化能和表面静电势
OH 自由基和丙烯的反应与乙烯的反应类似,但还是有不同的位点分析。如图7(a)所示,丙烯的ALIE极小值(8.89eV)出现在不饱和双键的中心,并不在饱和碳的位点,但此处也有淡蓝色区域,说明这两处都有可能是反应活性位点。如图7(b)所示,丙烯的ESP较小值出现在红色区域不饱和碳位置,此处的ESP 极小值为-0.72eV。而活性较弱的饱和碳处的ESP 值为0.08eV。虽然静电势是正值,但也有可能发生反应,因此对于此位点的反应,尝试了量子化学计算,并成功给出了过渡态结构,如图2 反应7 所示。OH 自由基进攻丙烯双键中心的反应过程与乙烯的类似,进攻时OH自由基裂解成氧自由基和氢自由基,氧自由基与丙烯结合成环氧丙烷中间体,反应路径如图2反应6所示。对于环氧丙烷本文也给出了活性位点,环氧丙烷的ALIE极小值也在氧原子处(11.53eV),ESP极小值也在氧原子处(-1.51eV),其他位置的ESP均没有氧原子处的小。因此结合ALIE 和ESP 分析来看,下一步的反应也会在氧原子处发生。
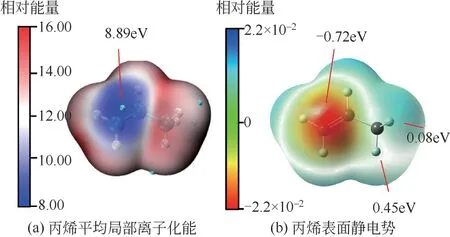
图7 丙烯平均局部离子化能和表面静电势
3.3 物种浓度及敏感性分析
通过对主要组分浓度的变化分析可知,柴油燃烧主要由前期的热解阶段和燃烧阶段两大阶段组成。Walton等通过实验证明了柴油具有两级着火的现象,因此本文量化计算所耦合的机理也很好地匹配了这一点。如图8(a)所示,在1.25ms附近,主要反应为低温区的裂解反应,正庚烷发生一定量的裂解,裂解反应会放出热量,导致系统温度有所升高,对应了途中曲线的温度台阶。虽然温度有所升高,但升高后的温度并不能促发甲苯的裂解,因为甲苯的苯环是一个共轭结构,要非常大的能量才能打破原子之间的共轭键,所以甲苯的物质的量分数并没有变化。而此时刻对应的二氧化碳有一定的上升,一氧化碳有一定的下降,这是因为二氧化碳具有第三体碰撞效应[CO+(M)—→— CO+O+(M)],一部分二氧化碳裂解为一氧化碳和氧自由基从而导致了二氧化碳的下降和一氧化碳上升。而1.5ms附近,是柴油着火时刻,正庚烷在1.5ms前已经燃烧殆尽,甲苯则是在1.5ms着火时刻燃烧殆尽,它们释放大量的热量,温度迅速升高,达到了二氧化碳热解的温度,因此图8(a)中一氧化碳曲线在此时刻有一个阶跃的上升。同时刻,正庚烷和甲苯的燃烧也会释放大量的二氧化碳,因为燃烧占主导部分,二氧化碳生成的量大于热裂解的量,导致了其有一定量的上升。因为此时刻是着火燃烧时刻,OH 自由基也有一定量的增加,这为第二着火阶段。图8(b)对点火时刻的OH 自由基敏感性分析来看,OH自由基对正庚烷链式反应中的产物(如CHO)呈正相关,说明升温提升反应速率,进而促进其燃烧,而对甲苯反应呈负相关,说明升温降低反应速率会阻碍其进程从而抑制燃烧,从图8(a)来看,甲苯的斜率比正庚烷的斜率小也印证了这一分析。

图8 物种浓度及反应敏感性
3.4 火焰自然光度与浮起长度
图9是在定容燃烧弹中六种工况下直接拍摄的火焰图像和处理的伪色图像,直接拍摄图像包含了更多的火焰局部特征,可以清楚地观察到火焰的真实形状。而利用火焰对应的伪色图像,可以更好地观察不同位置的发光强度。在柴油喷射到燃烧室后,热空气被夹带到腔内,火焰随着空气的夹带向外围发展,然后在锥形柴油束的外围形成稀薄的火焰前沿,并伴有火焰回溯现象,这与Dec和Curran等提出的碳烟火焰生成模型非常相似。但当工况为CO/O氛围时,其火焰结构与空气氛围下的结构不同,由于缺少氮气,火焰结构中并没有出现Dec提出的氮氧化物区域。随着二氧化碳浓度的提升,火焰发生分叉现象,湍流效果明显。但随着二氧化碳浓度比例的降低,火焰形状变得细长,湍流现象减弱,说明二氧化碳促使柴油燃烧火焰分叉和湍流。Pryor 等通过高速摄像图像和激光吸收光谱研究了添加二氧化碳对甲烷的燃烧。结果表明,大量二氧化碳的加入使得燃烧过程不再均匀,激波的分叉非常大。然而,他们只是研究二氧化碳对甲烷这种小分子燃烧的影响,对柴油大分子混合物是否产生类似的影响仍需深度研究。从伪色图像中可以看出,随着二氧化碳浓度的降低和氧气浓度的增加,着火时间从0.72ms 提前到0.49ms,这是二氧化碳和氧气的双重作用。此外,随着二氧化碳浓度降低,火焰回溯现象明显。在43%~35% CO体积分数范围,火焰回溯至喷嘴,说明此浓度范围有利于未燃烧烃类的消耗和燃烧效率的提高。可以看出,第一帧可见火焰在空气氛围下出现在0.72ms,最后一帧在1.70ms附近消失,火焰呈扁平状。当背景氛围是CO/O时,第一帧(最后一帧)的可见火焰出现时间分别为0.64ms(1.62ms)和0.50ms(1.48ms)。由图9观察到第一帧火焰是小而密集的条状,而最后一帧是不规则的形状,火焰的亮度比空气氛围下的弱。

图9 柴油燃烧火焰图像及其伪色图像
图10(a)为不同条件下的实验值,可以观察到空气氛围下的火焰浮起长度与CO/O氛围下火焰浮起长度有很大差异。总的来说,在空气中火焰浮起长度比在CO/O氛围中的要长,这是由于较高浓度的二氧化碳和氧气对柴油燃烧火焰的回溯现象有明显增强作用,导致了较短的火焰浮起长度。而在CO/O氛围下,火焰浮起长度随着二氧化碳浓度的降低和氧气浓度的增加而变短,因为氧气浓度的增加显著促进柴油燃烧。对于单一条件下的火焰浮起长度,无论是空气氛围还是CO/O氛围,随着火焰的传播,火焰浮起长度先变长,然后变得更短,最后变得更长。这是因为一开始火焰向下传播柴油喷射速率较高,所以会有较长的火焰浮起长度出现,但火焰立即回溯到喷嘴,浮起长度变短,实现稳定燃烧。而在柴油燃烧接近燃烧末期时,回溯现象不明显,较长火焰浮起长度出现。图10(b)~(d)为实验结果和定容燃烧弹实验仿真对比,如图10(b)所示,空气氛围下的火焰浮起长度和仿真之间存在一定误差,最大误差、最小误差和平均误差为分别为44.2%、5.9%和26.9%,因为该机理模型适用于CO/O气氛,消除了氮气对火焰浮起长度的影响,因此在空气氛围下拟合度不高。如图10(c)、(d)所示,仿真与CO/O氛围下的实验值吻合度较高。在图10(c)中,仿真与实验最大误差、最小误差和平均误差为分别为22.2%、0.6%和11.65%。在图10(d)中,仿真与实验最大误差、最小误差和平均误差分别为13.9%、0.5%和1.4%。因此,本工作提出的柴油燃烧量子化学计算机理在CO/O氛围下与实验值拟合度较高,对实际的闭式循环柴油机在CO/O氛围下的燃烧特性具有重要意义,对未来无氮排放、发动机制造技术和化工进展具有重要参考价值。
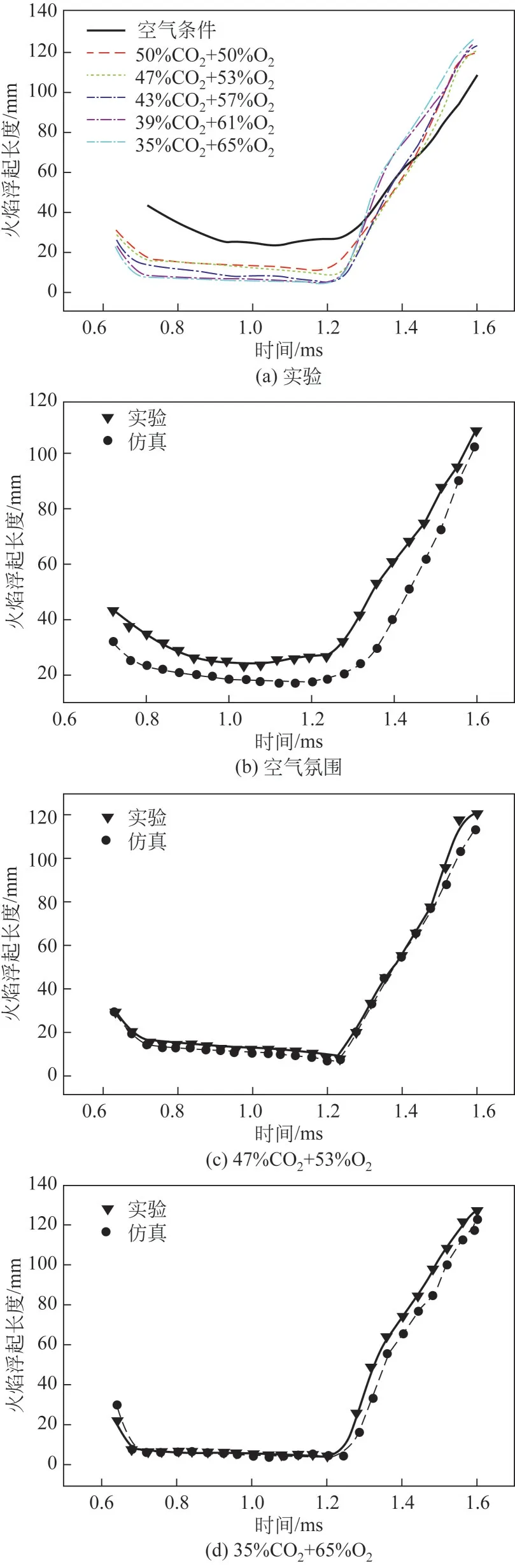
图10 火焰浮起长度实验值与仿真值
4 结论
为探究柴油在CO/O氛围,尤其是高浓度CO氛围下燃烧的特性,本文通过量子化学计算和化学反应动力学计算,提出新的化学反应路径,进一步搭建光学定容燃烧弹实验台架,进行定容燃烧弹可视化试验,将试验值与仿真值对比分析,主要得到以下结论。
(1)量子化学机理模型适用于研究柴油在CO/O氛围下的燃烧特性,其中在35%CO+65%O工况下火焰浮起长度最大误差、最小误差和平均误差分别为13.9%、0.5%和1.4%,均为可接受的误差,这项研究对未来无氮排放技术和闭式循环柴油发动机制造具有一定的参考价值。
(2)二氧化碳能够直接参与化学反应,且高温热解产物为一氧化碳与氢自由基。一氧化碳的碳原子端平均局部离子化能极小值为12.62eV,静电势极小值为-0.51eV。氧原子端的平均局部离子化能和静电势比碳原子端的大,因此一氧化碳的碳原子端活性比氧原子端活性大。
猜你喜欢
分析化学(2019年3期)2019-03-30
人人健康(2019年1期)2019-01-10
分析化学(2017年12期)2017-12-25
科学与财富(2017年27期)2017-10-17
百科知识(2016年16期)2016-10-29
