
钒前驱体对V-OMS-2结构及催化CO氧化活性的影响
2022-06-22 02:16汤清虎郭晓慧谢小培赵培正
河南师范大学学报(自然科学版) 2022年4期
汤清虎,郭晓慧,谢小培,赵培正
(河南师范大学 化学化工学院,河南 新乡 453007)
一氧化碳(CO)催化氧化因其在环境和工业领域的各种用途而被认为是一个重要的反应[1].尽管负载贵金属如Pt,Pd,Au等已被广泛用作消除CO的催化剂[2-4],但昂贵的价格和低的热稳定性制约了它们的实际应用.相比之下,负载或非负载过渡金属氧化物催化剂具有来源丰富、价格低廉、热稳定性高的特点,被认为是CO氧化的潜在候选催化剂[5-6].因此,研究过渡金属氧化物的物理化学性质并提高其催化性能具有重要科学和实际意义.
OMS-2是MnO2的同素异形体,是由锰氧八面体(MnO6)单元采用共棱角的联结方式构筑而成的2×2隧道结构,隧道开口约为0.46 nm.OMS-2中的Mn主要为以Mn4+离子形式存在,还有少量的Mn3+,Mn2+离子,平均氧化态为3.8价.OMS-2的混合价Mn离子、较大的外表面积及敞开的隧道结构使其在一些氧化反应中显现出较好的催化性能[7-10].在OMS-2中掺杂活性金属阳离子是常用的改善其结构和催化性能的策略.以往研究者的工作大多集中在低价金属阳离子掺杂OMS-2的研究上,而对高价金属阳离子(如M5+,M6+)的掺杂却少有报道[11].与低价金属阳离子相比,高价金属阳离子的酸性强、离子半径大,有利于OMS-2表面活性氧物种的形成.POLVEREJAN等[12]报道,在OMS-2骨架中掺杂V5+离子可加强其离子性,即增加了OMS-2中缺陷位的数目.TANG等[13]发现,少量V5+离子的掺杂显著提高了OMS-2对甲醛和甲烷低温燃烧的催化活性,掺杂V后,表面缺陷位(Lewis酸性)的增加以及氧化还原性的提高是OMS-2催化活性得以明显改善的主要原因.随后,GENUINO等[14]的研究工作显示,V掺杂量对OMS-2的结构及催化CO氧化反应活性具有显著的影响.为全面了解掺杂V对OMS-2结构及催化CO氧化的促进作用,本文采用简单回流法制备了不同钒前驱体掺杂的V-OMS-2催化剂,并研究了其对CO氧化的催化活性.为构建催化剂结构与活性的潜在联系,利用粉末X射线衍射(XRD)、氮气物理吸附(BET)、透射电镜(TEM)、X射线光电子能谱(XPS)、热重分析(TGA)、程序升温还原(H2-TPR)等实验技术对催化剂进行了表征.
1 实验部分
1.1 催化剂制备
采用回流法[14]制备未掺杂的OMS-2.将9.9 g MnSO4·H2O(分析纯)和3.4 mL HNO3(~68%,质量分数)溶解在35 mL二次水中制得溶液1.再将9.9 g KMnO4(分析纯)溶解在120 mL二次水中制得溶液2.将溶液2在不断搅拌下逐滴加入溶液1中.可观察到逐渐有棕黑色沉淀物生成,将该混合液回流24 h.自然冷却后,将棕黑色沉淀物过滤、洗涤,并在110 ℃鼓风烘箱中干燥12 h,而后在250 ℃下煅烧3 h,即得未掺杂的OMS-2.为获得V掺杂的OMS-2样品,在溶液2滴加完后,再加入一定量的五氧化二钒(V2O5,分析纯)或偏钒酸钠(NaVO3,分析纯),其他制备步骤与未掺杂的OMS-2相同.将所制得的V掺杂OMS-2催化剂标记为x%V-OMS-2(y),其中x%为V掺杂OMS-2催化剂中V与Mn的原子百分比,y表示V2O5(y=1)或NaVO3(y=2).
为了便于比较,还采用湿法浸渍法制备了3%V/OMS-2催化剂.将2.0 g未掺杂的OMS-2加入偏钒酸铵(约10 mL)水溶液中,连续搅拌2 h后静置24 h.将混合液放入80 ℃水浴中加热,蒸去多余的水.将残余的固体物放入120 ℃鼓风烘箱中干燥10 h,而后转入250 ℃马弗炉中煅烧3 h,制得的浸渍V催化剂样品标记为3%V/OMS-2.
1.2 催化剂表征
未掺杂和V掺杂OMS-2样品的晶相分析在Bruker D8 Advance 型粉末X射线衍射仪上进行.各样品的比表面积和孔径分布在Micromeritics ASAP 2020型物理吸附仪上测定.使用JEOL JEM-2100型电子显微镜观测各样品的形貌和晶格相.采用KBr压片法,在Nicolet Nexus型红外光谱仪上测定各样品的FT-IR谱.在Perkin Elmer ELAN DRC-e型电感耦合等离子体质谱仪(ICP-MS)上测定样品中各元素的含量.在Thermo Fisher ESCABALE 250Xi型能谱仪上记录各样品的X射线光电子能谱.各样品的热分析曲线在Netzsch STA449C型综合热分析仪上测定.各样品的氧化还原性能在自组装的带有自动升温程序和TCD检测器的流动系统中测定.称取20 mg样品装入石英反应管中,通入干燥的空气,在250 ℃下处理1 h,而后切换为纯N2(总流量均为30 mL/min).当反应管温度降至50 ℃以下时,切换含5% (体积分数)H2和95%(体积分数)Ar的混合气(总流量为30 mL/min),以10 ℃/min的升温速率测定各样品的H2-TPR曲线.
1.3 催化剂活性评价
未掺杂和V掺杂OMS-2的CO氧化活性在石英管(内径9 mm)固定床反应器中测定.称取100 mg催化剂装入石英反应管中,通入干燥的空气,在250 ℃下处理1 h.当反应管温度降至30 ℃以下时,切换含1%(体积分数)CO和99 %(体积分数)空气的混合气(总流量为50 mL/min)开始反应,反应温度由放置在催化剂床层间的热电偶监测.利用甲烷转化器将通过催化剂床层后混合气中的CO和CO2全部转化为甲烷,并由带有氢火焰离子化(FID)检测器的GC-9160型气相色谱仪(上海欧华分析仪器厂)在线分析.
2 结果与讨论
2.1 钒掺杂量的优化
从图1(a)看出,当V与Mn原子比不高于3%时,V-OMS-2(1)仍能保持OMS-2的晶相结构,但衍射峰强度逐渐减弱;而当V与Mn原子比高于6%时,仅在2θ=38°处呈现一归因于非晶态水钠锰矿型MnO2(JCPDS 42-1317)的较宽衍射峰,这可能是由于掺杂的V阻碍了OMS-2晶相的形成所造成的.未掺杂,1%,3%,6%,12%V掺杂OMS-2的CO转化随温度变化曲线见图1(b).与未掺杂OMS-2相比,所有V掺杂OMS-2均呈现较高的CO氧化活性.其中,3%V-OMS-2(1)的活性最高.该催化剂的t50%(CO转化率为50%的温度)为22 ℃,并按以下顺序递增:3%V,6%V,1%V,12%V,未掺杂OMS-2(见表1).尽管6%V-OMS-2(1)具有更大的比表面积(241 m2/g),但其催化活性并不是最高的,说明保持OMS-2特有的晶相结构对催化CO氧化也是很重要的.可见,3%V为最佳的V掺杂量.
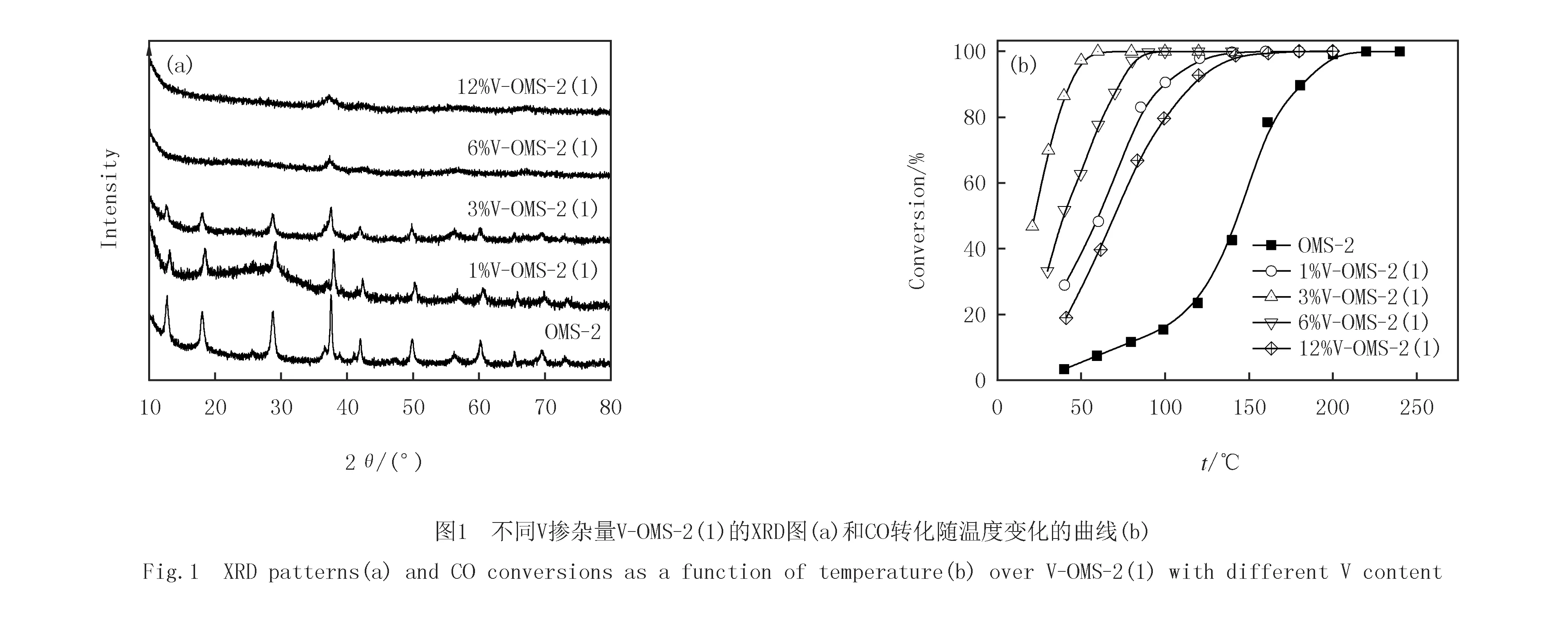
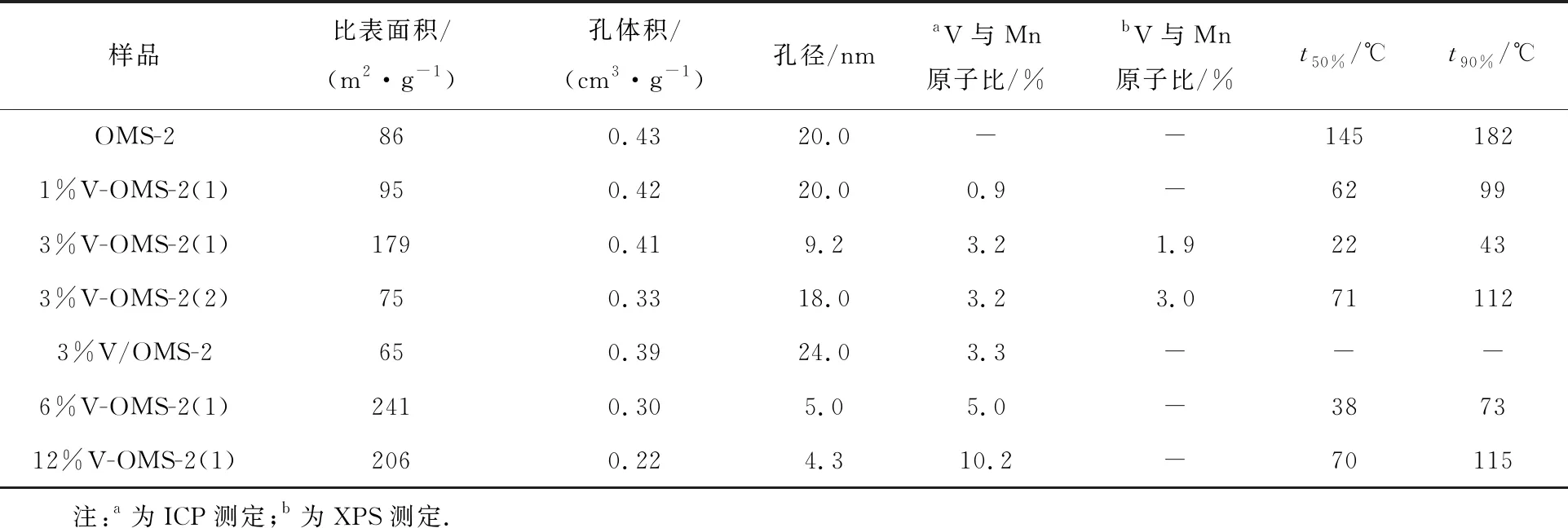
表1 未掺杂和V掺杂OMS-2的物理化学性质及催化性能
2.2 不同钒前驱体掺杂3%V-OMS-2的结构及催化活性

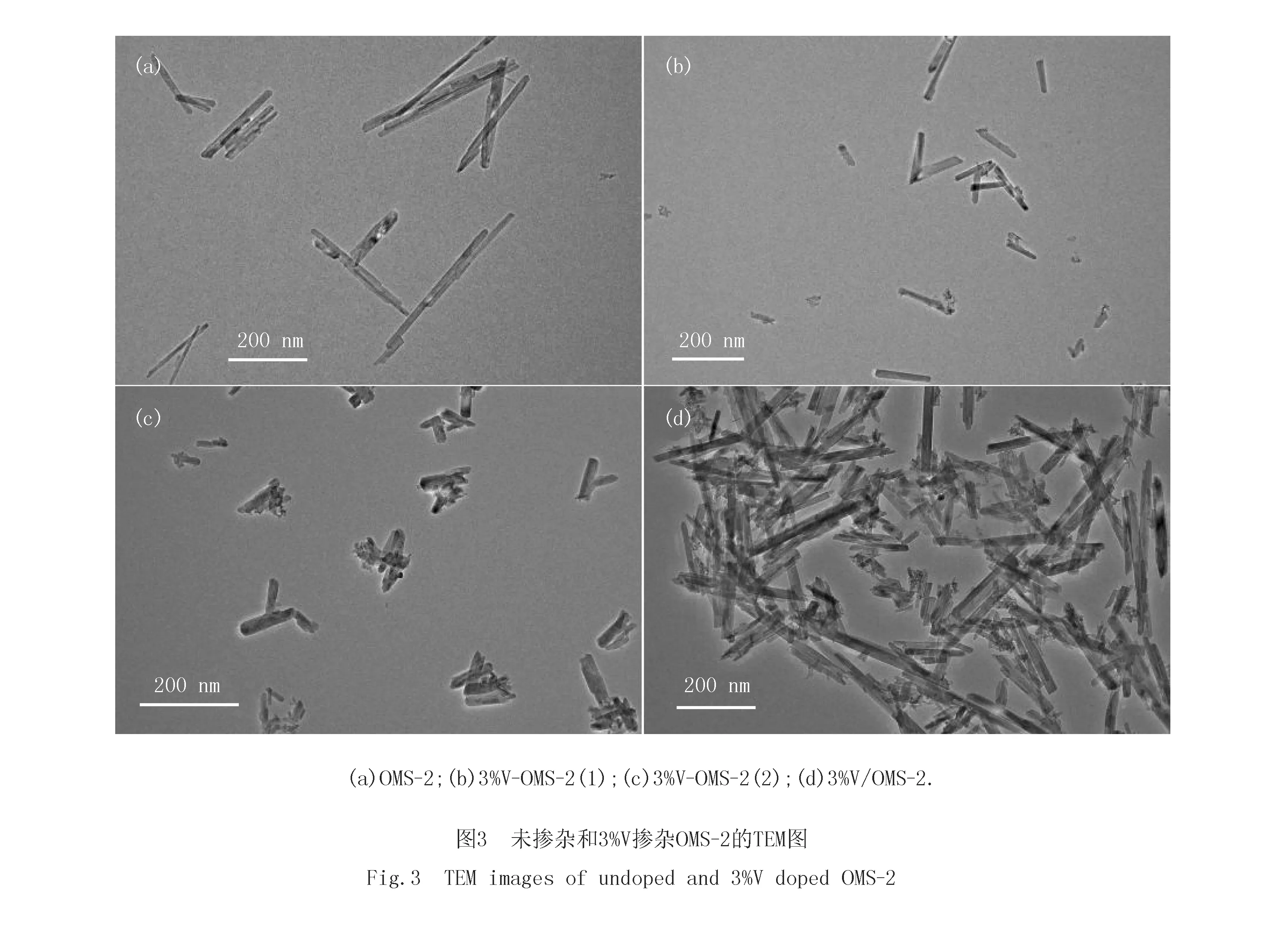
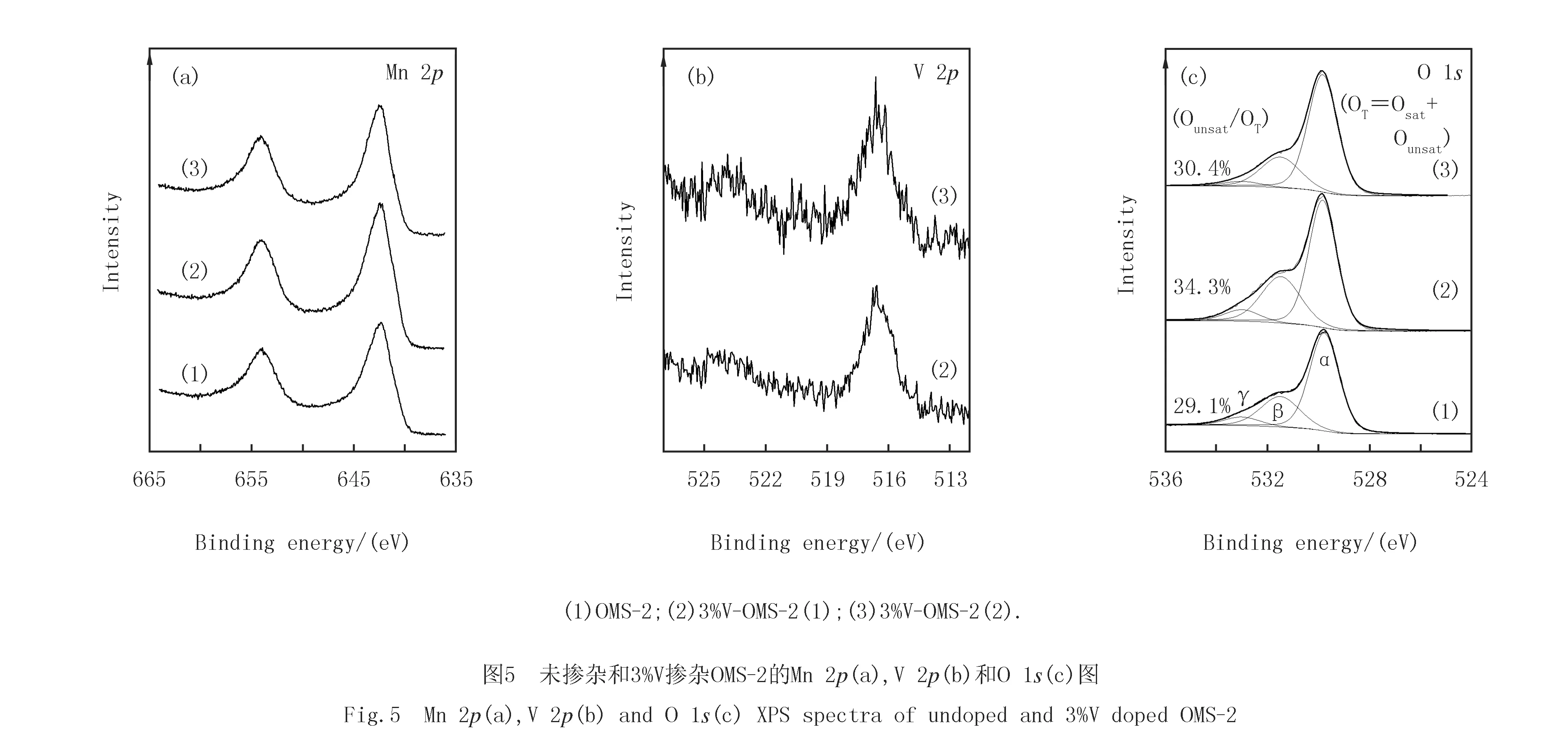
图3为未掺杂OMS-2和不同钒前驱体掺杂3%V-OMS-2的TEM图.可见,未掺杂OMS-2呈纳米棒状形貌(见图3(a)).浸渍V的3%V/OMS-2除部分发生断裂和团聚外,基本保持了OMS-2的原始形貌(见图3(d)).尽管3%V-OMS-2(1)和3%V-OMS-2(2)也呈纳米棒形貌(见图3(b)和3(c)),但纳米棒的平均长度却由未掺杂OMS-2的200~400 nm缩短到50~100 nm,而平均宽度则由原来的约10 nm增加到约20 nm,形成短纳米棒形貌.值得注意的是,所有3%V掺杂OMS-2的TEM图中均未观测到其他的分离相.这一现象进一步证实,掺杂的V主要以高分散态形式存在或置换Mn4+离子进入OMS-2的骨架中.
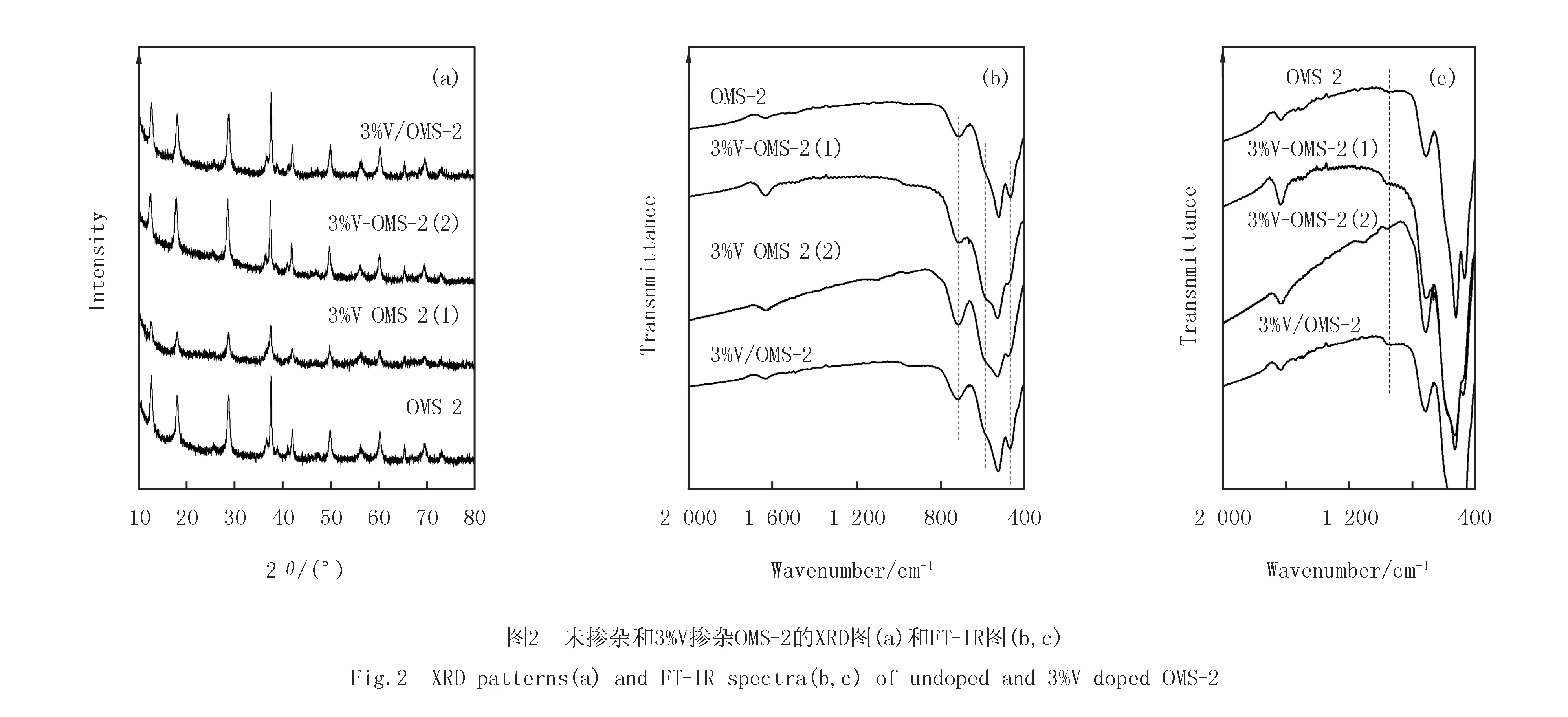
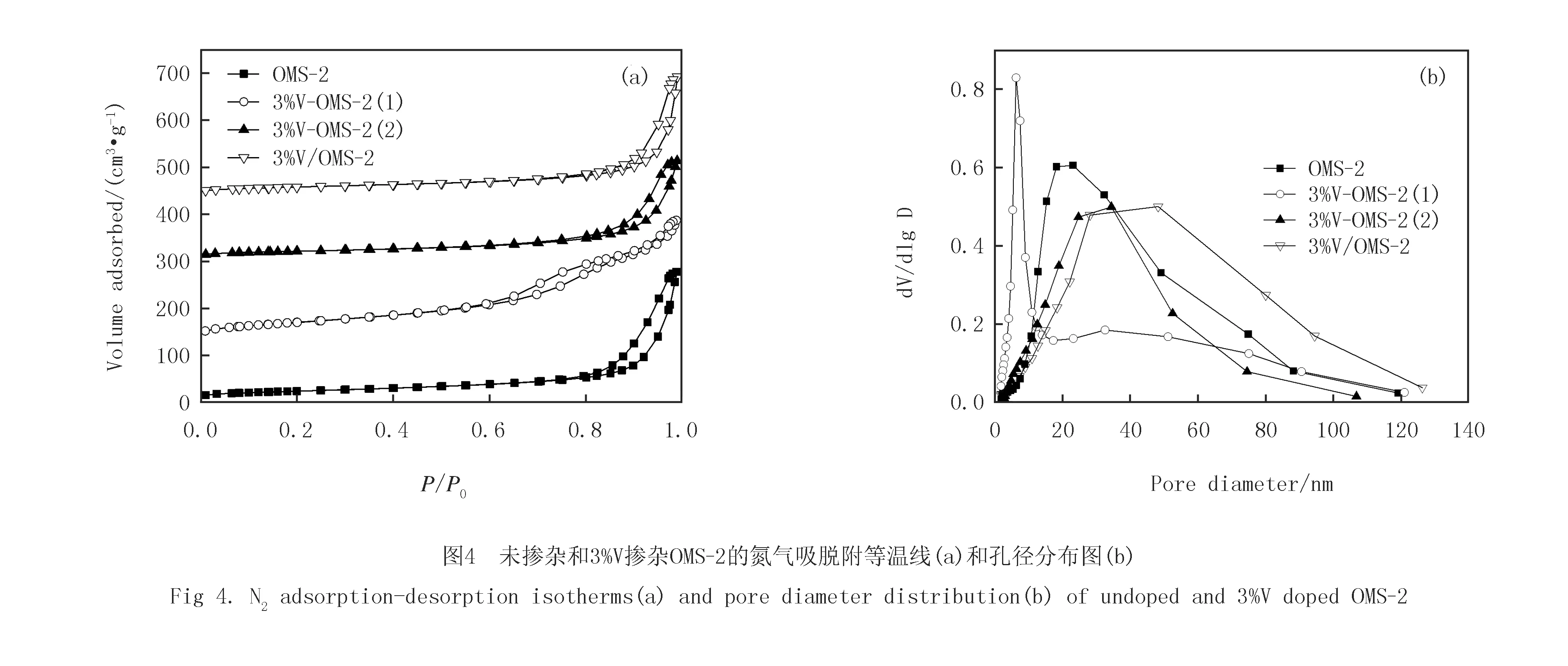
未掺杂和3%V掺杂OMS-2均呈Ⅱ型吸附等温线(见图4(a)),对应的BJH孔径分布曲线如图4(b)所示.由表1看出,未掺杂OMS-2的平均孔径为20.0 nm,比表面积和孔体积分别为86 m2/g和0.43 cm3/g.与未掺杂的OMS-2相比,3%V/OMS-2和3%V-OMS-2(2)的平均孔径改变不大,但比表面积和孔体积有所减小.对于以V2O5为前驱体掺杂的3%V-OMS-2(1),其吸脱附等温线的滞回环明显向低压区移动,且孔径分布变窄,平均孔径减小(20.0→9.2 nm),比表面积显著增加(86→179 m2/g),而孔体积仅略有下降(0.43→0.41 cm3/g).
未掺杂和3%V掺杂OMS-2的热分析曲线(见图6(a))类似,均呈现3个主要的失重温度带.其中,30~250 ℃的失重带为表面物理吸附的H2O和O2,随后是化学吸附H2O的脱附;第2个主要失重带发生在580~600 ℃,主要为表面吸附氧的释出[14];第3个主要失重温度带发生在710~750 ℃,归因于KMn8O16→ Mn2O3→Mn3O4晶相转变所引起的晶格氧的释出[17].未掺杂OMS-2和3%V-OMS-2(2)从(1)到(3)的失重曲线接近,只是3%V-OMS-2(2)在低温区的斜率略陡峭,第2个失重带的温度稍低.值得注意的是,以V2O5为前驱体掺杂3%V-OMS-2(1)在低温区具有更陡峭的斜率,暗示该样品在未发生相变前可释放出大量的物理和化学吸附H2O和O2.与未掺杂OMS-2和3%V-OMS-2(2)相比,3%V-OMS-2(1)第2个主要失重带的温度明显向低温偏移,说明其中的表面吸附氧更易释出,这预示着该样品中表面吸附氧的机动性、反应性更强.
采用H2-TPR技术测定了未掺杂和3%V掺杂OMS-2的氧化还原性能(见图6(b)).未掺杂OMS-2在250~400 ℃范围内发生还原,可分为α(323 ℃),β(359 ℃)和γ(376 ℃)3个还原峰.其中,较低温度的α峰归因于表面吸附氧的还原[22].随后的两个还原峰(β峰和γ峰)则归因于MnO2→Mn3O4→MnO的分步还原[23].3%V-OMS-2(2)的还原曲线与未掺杂OMS-2类似,只是α峰温度稍低,β峰和γ峰温度略高.与未掺杂OMS-2相比,以V2O5为前驱体掺杂3%V-OMS-2(1)的α还原峰从323 ℃降至295 ℃,β还原峰也从359 ℃降至321 ℃,说明3%V-OMS-2(1)更易于发生还原.一般来说,氧化物催化剂的较低还原温度意味着更高的氧物种迁移速率[24].因此,掺杂V5+置换Mn4+加强了结构氧在OMS-2表面的迁移[14].这与TGA和XPS的结果一致,说明以V2O5为前驱体掺杂V激活了OMS-2中的Mn-O键,形成了更多机动性强、更活泼的表面氧物种.相比之下,浸渍V的3%V/OMS-2的还原延迟至350~500 ℃范围内.考虑到钒氧物种的还原一般发生在500 ℃[25]以上,造成这一现象的可能原因是位于OMS-2外表面的钒氧物种妨碍了OMS-2中Mn离子的还原.
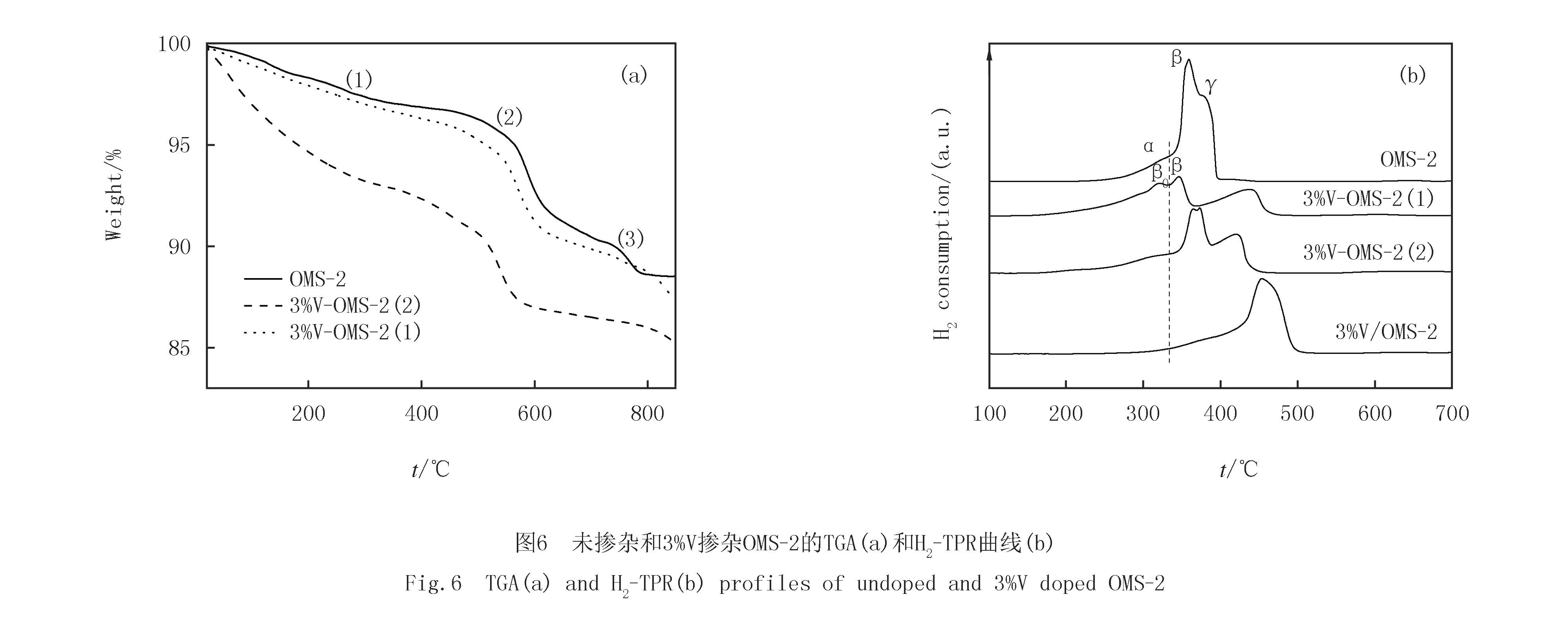
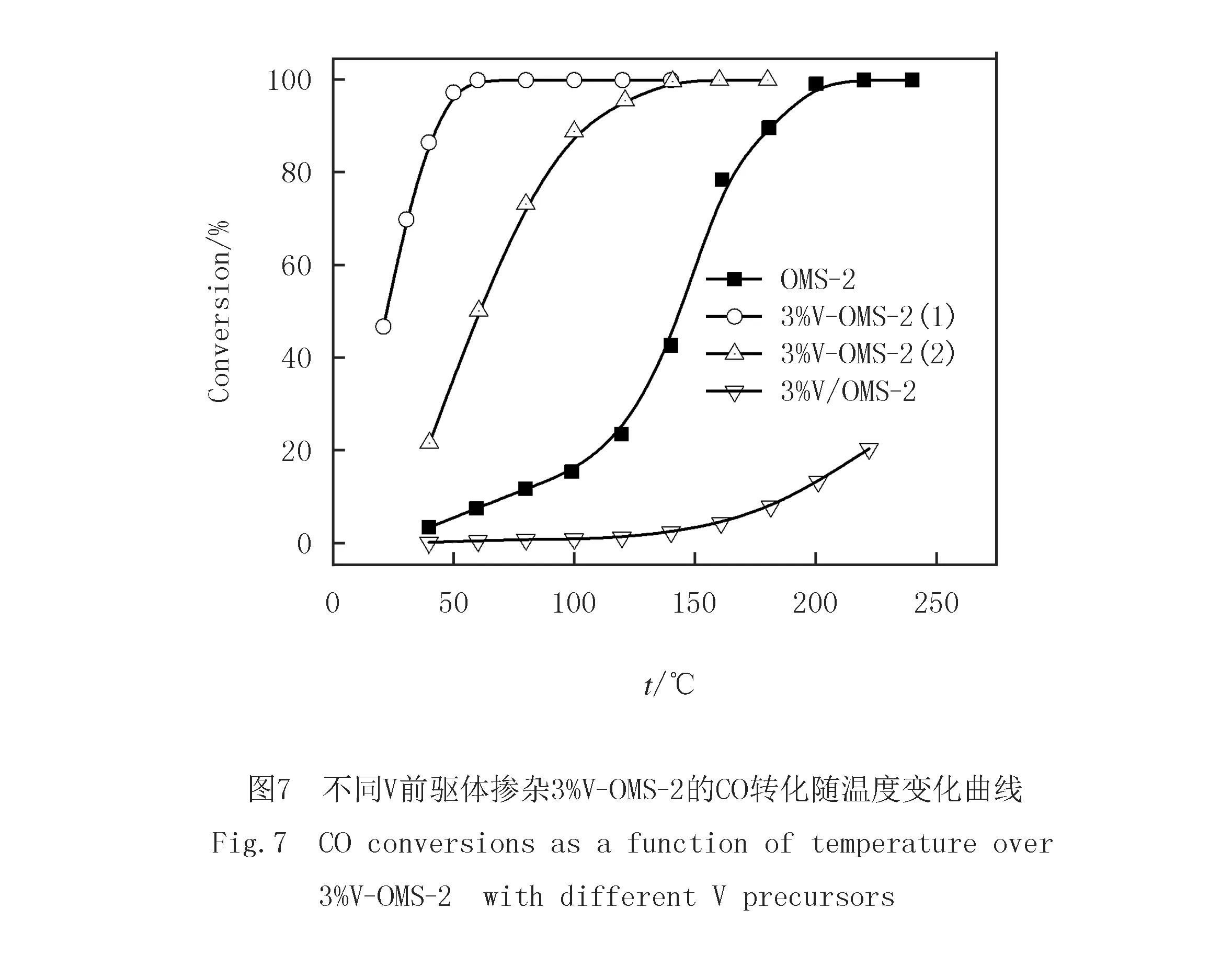
图7比较了未掺杂和不同钒前驱体3%V掺杂OMS-2上的CO氧化活性.与未掺杂OMS-2相比,3%V/OMS-2(浸渍3%V催化剂)的活性明显降低,说明负载的V对CO氧化无明显促进作用.位于OMS-2表面的负载V物种阻碍了CO分子在Mn活性位上的活化吸附,这可能是导致浸渍V催化剂活性下降的重要原因.采用回流法制备的3%V掺杂OMS-2催化剂均呈现出远高于未掺杂OMS-2的CO氧化活性.t50%按以下次序递增: 3%V-OMS-2(1),3%V-OMS-2(2),未掺杂OMS-2.其中,以V2O5为前驱体掺杂3%V-OMS-2(1)活性最高,可将CO完全转化温度由未掺杂OMS-2的200 ℃降至50 ℃以下.显然,不同钒前驱体对掺杂3%V-OMS-2催化CO氧化性能有重要影响.
文献[14,26]报道显示,OMS-2上的CO氧化遵循Mars-van Krevelen机理:OMS-2的表面吸附氧可在不破坏OMS-2的结构前提下作为氧化剂氧化CO;反过来,分子氧表面再吸附的易翻转性保证了OMS-2在催化CO氧化反应中的持久性.因此,OMS-2表面吸附氧的数量、机动性及氧化还原性是影响催化CO氧化反应的重要因素.XRD,TEM和H2-TPR结果表明,采用回流法掺杂3%V时,能够在保持OMS-2结构的前提下,改变OMS-2的形貌,增加缺陷位(吸附位点)的数量,并加强表面吸附氧的机动性和反应性,从而显著提高了其在CO氧化反应中的催化活性.XPS和H2-TPR结果显示,以V2O5为前驱体掺杂V制备的3%V-OMS-2(1)具有更多的表面吸附氧和较低的还原温度,这使其呈现更高CO氧化催化活性的重要原因.SUN等[27]报道,催化剂大的比表面积也有助于催化氧化反应的进行.由表1看出,以V2O5为前驱体掺杂V制备的3%V-OMS-2(1)具有更大表面积,这是其表现出较高催化活性的另一重要原因.
3 结 论
除浸渍V样品外,掺杂V均能显著改善OMS-2催化CO氧化反应的活性,这是由于掺杂的V5+离子置换Mn4+离子进入OMS-2的骨架中使锰氧八面体配位氧的环境发生了变化,从而改变了OMS-2的形貌,增加了表面缺陷位的数量,提高了表面活性氧的机动性和反应性.与其他钒前驱体掺杂的OMS-2相比,以V2O5为前驱体掺杂V制备的3%V-OMS-2(1)具有更高的催化活性,这与其较大的比表面积、更多数量机动性、反应性强的表面吸附氧及其较好的氧化还原性有关.
猜你喜欢
小学生学习指导(高年级)(2022年3期)2022-03-29
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
初中生世界·七年级(2021年2期)2021-03-12
证券市场周刊(2020年46期)2020-12-28
装备维修技术(2020年5期)2020-11-20
牡丹江师范学院学报(自然科学版)(2019年2期)2019-09-10
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
汽车与运动(2017年4期)2017-07-16
小学生导刊(高年级)(2017年2期)2017-06-10
