
玻璃珠-涡旋振荡改良法用于硅藻DNA 提取
2022-06-21 01:23:38蔡杰王博胡孙林曲一泓宋涛陈建华邓建强
法医学杂志 2022年1期
蔡杰,王博,胡孙林,曲一泓,宋涛,陈建华,邓建强
1.海南医学院法医学教研室 海南省热带法医学司法鉴定工程研究中心,海南 海口 571199;2.深圳美恩美科技有限公司,广东 深圳 518103
水中尸体的鉴定一直是法医学领域关注的问题之一,其中生前溺水与死后抛尸入水的鉴别依然是当前所面对的重要难题[1],传统依赖尸体征象的病理学检验特异性有限且受死亡时间、尸体腐败等条件影响大,因此,针对水中尸体组织器官中硅藻含量的检验,目前被认为是辅助溺死鉴定的“金标准”[2]。硅藻的检验主要分为形态学方法及分子生物学方法,其中形态学检验会受生物发育阶段和遗传可变的影响而产生变异,且硅藻种类过多,基于形态学的硅藻鉴定难度较大[3];而硅藻分子生物学检验方法具有无需硝酸消化、检验速度快、分类不依赖形态学特征,灵敏度、特异性和检出率更高,并可有效减少外来污染的影响,可依靠基因组数据库进行结果数据分析,如在人体器官中检出硅藻,是判断生前溺死的重要依据[4-7]。硅藻分子检验中,由于硅藻结构的特殊性[如硅藻细胞外覆主要成分为二氧化硅(SiO2)的硅质细胞壁],一般认为普通DNA 提取试剂盒无法破坏硅藻坚硬的硅质外壳[8-9],因此硅藻DNA 提取比较困难。同时,从人体组织中提取硅藻成分目前主要用于法医学实践,应用局限,因而不受试剂盒开发商关注,且其提取会受到人体组织的干扰,比水样硅藻DNA 提取的难度更大。因此,目前市面上针对法医学鉴定的硅藻提取试剂盒较少,多选用土壤DNA 提取试剂盒作为法医学硅藻DNA 检验的标准技术方案,也有采用植物DNA 提取试剂盒[10-12]。本研究选用2 种常用的基于不同技术原理的植物DNA 提取试剂盒(新型植物基因组DNA 提取试剂盒、Plant DNA Isolation 试剂盒)和1 种血液DNA 提取试剂盒(全血基因组DNA 提取试剂盒)提取硅藻DNA,在提取过程中加入玻璃珠和涡旋振荡进行改良,以目前使用率较高的DNeasy PowerSoil Pro试剂盒[10,13-14]为对照,检验该改良方法在提高法医学硅藻DNA 提取效果中的价值。
1 材料与方法
1.1 材料
1.1.1 样本
经确定为生前溺死者的肺组织1例及溺死区域水样,未经甲醛处理。
1.1.2 主要仪器和试剂
MIX-30S 迷你混合仪(杭州米欧仪器有限公司),SCILOGEX D3024 离心机(美国SCILOGEX 公司),Heme 9600 基因扩增仪(珠海黑马医学仪器有限公司),BG-subMIDI 电泳仪(北京百晶生物技术有限公司),Tanon 3500R 凝胶成像仪(上海天能科技有限公司),Rotor-Gene Q5PlexHRM 高分辨荧光定量PCR 仪(德国Qiagen 公司)。
分散研磨专用玻璃珠直径为1.5~2.0 mm(以下简称“大玻璃珠”)及0.4~0.6 mm(以下简称“小玻璃珠”),购自上海颖旭化工机械有限公司;DNeasy PowerSoil Pro 试剂盒(以下简称“PowerSoil 试剂盒”)购自德国Qiagen 公司;新型植物基因组DNA 提取试剂盒(以下简称“TIANGEN 试剂盒”)购自北京天根生化科技有限公司;Plant DNA Isolation 试剂盒(以下简称“FOREGENE 试剂盒”)购自成都福际生物技术有限公司;全血基因组DNA 提取试剂盒(以下简称“BioTeKe 试剂盒”)购自北京百泰克生物技术有限公司;PCR Mix 购自北京中科瑞泰生物科技有限公司;MonAmpTMChemoHS qPCR Mix 购自莫纳生物科技有限公司。引物由北京中美泰和生物技术有限公司合成。
1.2 方法
1.2.1 DNA 提取改良条件的确定
DNA 提取效果受不同大小玻璃珠的比例和涡旋振荡时间影响[15],在涡旋振荡频率为3 000 r/min 的条件下,为获得提取DNA 的最优条件,设计不同大小玻璃珠混合比例和涡旋振荡时间组合(表1),使用FOREGENE 试剂盒对组织中硅藻DNA 进行提取,对提取产物直接进行电泳,并应用藻类特异性引物进行PCR 扩增后电泳。重复实验3 次。以电泳条带为评价指标,相同涡旋振荡时间内较亮条带确定不同玻璃珠混合的最优比例,不同振荡时间电泳条带稳定出现的最短时间为最佳时间。
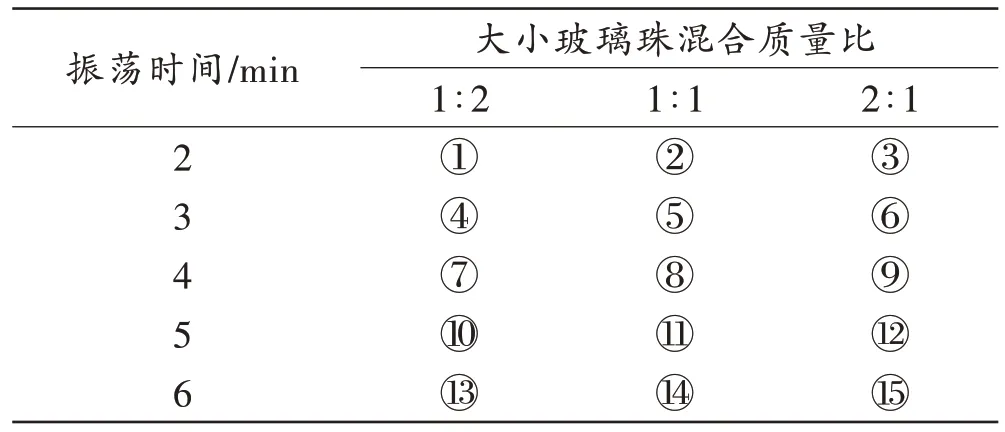
表1 不同玻璃珠比例和涡旋振荡时间的组合Tab.1 Combination of different glass bead proportion and vortex oscillation time
1.2.2 样本前处理
由于送检案件肺组织和水样量的限制,本研究4 种试剂盒均采取水样和组织样本各5 份进行实验。以PowerSoil 试剂盒作为对照组,另外3 种试剂盒分为按说明书操作的常规组3 组以及改良组3 组。7 组共70 份样本。每份水样取10 mL,13 400×g离心5 min,弃去上清液,留置底部液体200 μL 待检。每份取肺边缘组织0.5 g,充分剪碎。
1.2.3 DNA 提取方法
1.2.3.1 对照组
严格按照PowerSoil 试剂盒提供的操作说明书[12],分别对水样及组织进行DNA 提取,共计10 份对照样本。具体操作步骤如下:取充分剪碎的肺组织0.5 g加入含玻璃珠的离心管中,加入800 μL CD1 溶液,涡旋振荡15 s 充分混匀,将含玻璃珠的离心管插入MIX-30S迷你混合仪的涡旋适配器上涡旋振荡10 min,然后15 000×g离心1 min,转移上清液600 μL 至2 mL离心管中,添加200 μ L CD2 溶液涡旋振荡5 s,15 000×g离心1 min。取600 μL 上清液加入2 mL 离心管内,再加入600 μL CD3溶液涡旋振荡5 s,取650 μL上清液转移至旋转离心柱中,15 000×g离心1 min,弃去下方废液,将剩余液体加入旋转离心柱,15 000×g离心1 min 后倒掉废液,再将旋转离心柱置于新的2 mL 离心管中,加 入500 μL EA 溶液15 000×g离心1 min,弃去废液后放回离心管。加入500 μL C5 溶液,15 000×g离心1 min,将旋转离心柱置于新的2 mL 离心管中,16 000×g离心2 min,将旋转离心柱置于1.5 mL 洗脱柱中,加100 μL C6 溶液到旋转离心柱中心白色过滤膜上,15 000×g离心1 min,弃去旋转离心柱,此时收集管中的DNA 用于后续实验。
1.2.3.2 常规组
分别选用TIANGEN 试剂盒、FOREGENE 试剂盒、BioTeKe 试剂盒进行DNA 提取。严格按照3 种试剂盒的说明书[16-17]提取水样和组织的DNA,每个试剂盒均提取水样和组织DNA各5份,3种试剂盒共计30份实验样本。
(1)TIANGEN试剂盒。取肺组织0.5 g,加入400 μL缓冲液LP1 和6 μL Rnase A(10 mg/mL),旋涡振荡1 min,室温放置10 min。加入130 μL 缓冲液LP2,充分混匀,涡旋振荡1 min,13 400×g离心5 min,将上清液移至新的离心管中,然后加入1.5 倍体积的缓冲液LP3,立即充分振荡混匀15 s,此时可出现絮状沉淀。将上一步所得溶液和絮状沉淀都加入1 个吸附柱CB3 中,吸附柱放入收集管中,13 400×g离心30 s,弃去废液,将吸附柱CB3 放入收集管中。向吸附柱CB3中加入600 μL 漂洗液PW,13 400×g离心30 s,弃去废液,将吸附柱CB3 放入收集管中,再次加入600 μL 漂洗液PW 后13 400×g离心30 s,弃去废液,将吸附柱CB3 放回收集管中,13 400×g 离心2 min,弃去废液。将吸附柱CB3 置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液。最后将吸附柱CB3 转入1 个干净的离心管中,向吸附膜中央滴加100 μL 洗脱缓冲液TE,静置5 min,13 400×g离心2 min,离心管中的液体即为提取的DNA 溶液。
(2)FOREGENE 试剂盒。将600 μL 缓冲液PL1、20 μL Foregene 蛋白酶、2 μL β-巯基乙醇加入2 mL离心管中,混匀后65 ℃预热,将0.5 g 肺组织加入离心管内迅速颠倒混匀,水浴10 min,每隔5 min 颠倒混匀1 次。加入600 μL 缓冲液PL2 充分混匀,继续置于65 ℃ 10 min,然后13 400×g离心5 min,将上清液转移至新的离心管中,加入180 μL 无水乙醇,涡旋振荡15 s充分混匀,此时可出现絮状沉淀。将离心柱放入收集管中,取800 μL 混合液加入离心柱,13 400×g离心1 min,弃去废液,将离心柱放回收集管中,将剩余混合液全部加入离心柱中,13 400×g离心1 min,弃去废液。再向离心柱中加入500 mL漂洗液PW,13 400×g离心30 s后弃去废液。向离心柱中加入700 μL漂洗液WB,13 400×g离心30 s,弃去废液,再重复加入700 μL漂洗液WB,13 400×g离心30 s,弃废液。将离心柱放回收集管中,13 400×g空管离心2 min,将离心柱转移至新的1.5 mL 离心管中,向膜中央悬空滴加100 μL已于65 ℃预热的洗脱液EB,室温放置2 min,13 400×g离心1 min。再次向胶膜中央悬空滴加100 μL 已预热的洗脱液EB,13 400×g离心1 min。将2 次收集的洗脱液汇集即完成DNA 提取。
(3)BioTeKe 试剂盒。向吸附柱AC 中(吸附柱放入收集管中)加入500 μL 平衡液,13 400×g离心1 min,弃去收集管中的下滤液,将吸附柱重新放回收集管中。加入40 μL PK(20 mg/mL),室温放置15 min,颠倒混匀几次,加入200 μL 结合液CB,立刻涡旋15 s混匀,70 ℃放置10 min,变黑色清亮溶液。加入100 μL异丙醇,涡旋15 s 混匀,此时可出现絮状沉淀。将上一步所得溶液和絮状沉淀都加入一个进口吸附柱AC中,吸附柱放入收集管内,8 000×g离心30 s,弃去收集管中的废液。加入500 μL 抑制物去除液IR,13 400×g离心30 s,弃去废液,加入700 μL 漂洗液WB,13 400×g离心30 s,弃去废液,加入500 μL 漂洗液WB,13 400×g离心30 s,弃去废液。将进口吸附柱AC 放入空收集管中,14 500×g离心2 min,尽量除去漂洗液。取出进口吸附柱AC,置于干净的离心管中,在吸附膜的中间部位加100 μL 洗脱缓冲液EB,室温放置3~5 min,13 400×g离心1 min。将得到的溶液重新加入离心吸附柱中,静置2 min,13 400×g离心1 min,即为提取的DNA 溶液。
1.2.3.3 改良组
根据1.2.1 节实验所得最佳玻璃珠和涡旋振荡时间的组合条件,将固定组分的玻璃珠随组织同时加入2 mL 微量离心管中,实验过程中的DNA 消化步骤按3 000 r/min 4 min的条件进行涡旋振荡,如FOREGENE试剂盒和BioTeKe 试剂盒均为加入蛋白酶后振荡4 min,TIANGEN 试剂盒为加入缓冲液LP1 后振荡4 min。其他实验步骤严格按照试剂盒说明书进行操作。每种试剂盒均提取水样和组织DNA各5份,3种试剂盒共计30 份实验样本。
1.2.4 DNA 提取产物的检测和验证
1.2.4.1 琼脂糖凝胶电泳
各组DNA 提取产物直接通过2%琼脂糖凝胶进行电泳,在Tanon 3500R凝胶成像仪下进行观察和分析。
1.2.4.2 PCR 和电泳
选取硅藻特异性引物,将4 种试剂盒共70 份提取物进行PCR 扩增,以确定DNA 提取产物中是否含有硅藻DNA 成分。
选用ZIMMERMANN 等[18]研究的可特异性扩增硅藻18S rDNA 片段的引物,应用Basic Local Alignment Search Tool(BLAST,https://blast.ncbi.nlm.nih.gov/Blast.cgi)对引物的硅藻特异性进行检验,同时排除该引物扩增人类基因组的可能。引物序列为18S 正向引物D512(5′-ATTCCAGCTCCAATAGCG-3′)、18S 反向引物D978(5′-GACTACGATGGTATCTAATC-3′),扩增长度为390~410 bp。PCR 反应总体积为25 μL,包括模板DNA 2 μL,正反向引物(10 μmol/L)各0.5 μL,ddH2O 17.75 μL,10×缓冲液2.5 μL,脱氧核糖核苷三磷酸(deoxy-ribonucleo-side triphosphate,dNTP)0.5 μL,再加入Taq酶1.25 μL,空白对照将模板DNA 换为等量ddH2O。扩增条件:94 ℃ 10 min;94 ℃ 45 s,50 ℃45 s,72 ℃1 min,40 个循环。
所有样品的PCR 扩增产物通过2%琼脂糖凝胶进行电泳,电泳后Goldview 显色,使用Tanon 3500R凝胶成像仪观察和分析。
1.2.5 qPCR 产物定量及数据分析
qPCR反应总体积为20 μL,包括模板DNA 2 μL,正反向引物(10 μmol/L)各0.4 μL,ddH2O 7.2 μL,qPCR mix 10 μL。扩增条件:94 ℃10 min;94 ℃45 s,50 ℃45 s,72 ℃1 min,40 个循环。
在实时荧光PCR 中,每个模板的Ct 值与该模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,Ct 值越小[19],可通过qPCR 得到改良前后的Ct 值。数据采用均值±标准差()表示,采用SPSS 17.0 软件(美国SPSS 公司)对数据进行方差分析,组间比较使用最小显著差异法(least-significant difference,LSD)-t检验。检验水准α=0.05。
2 结果
2.1 引物的特异性检验
BLAST 检验结果显示,18S rDNA 的引物D512、D978 为藻类特异性引物,扩增结果不显示人类基因。
2.2 DNA 提取的最优条件
将不同条件下提取的DNA 样本直接进行电泳,各组均未检出DNA 条带。但将不同条件下提取的DNA 进行硅藻特异性PCR 扩增后电泳,均获得了特异性的硅藻18S rDNA 扩增条带,提示本研究所设计的DNA 提取方法成功提取到硅藻DNA。
经3 次重复性实验确证,在3 000 r/min 涡旋振荡4 min 条件下,电泳条带亮度趋于稳定;在相同涡旋振荡时间下,⑤、⑧、⑭3 个条带较亮,即当2 种规格玻璃珠按1∶1 的质量比混合时,电泳条带亮度亮于其他组合(图1)。
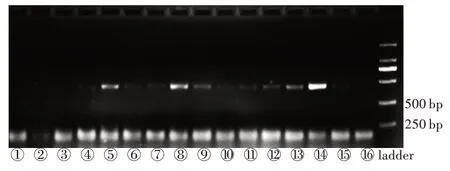
图1 不同玻璃珠比例和涡旋振荡时间组合的电泳结果Fig.1 The electrophoresis result of different glass bead mass proportions and vortex oscillation time combinations
2.3 PCR 和电泳
和试剂盒的常规操作效果比较,采用玻璃珠-涡旋振荡改良后,用于植物提取的2 种试剂盒(TIANGEN 和FOREGENE 试剂盒)提取水样中硅藻DNA 的电泳条带亮度增加,而组织样本亮度变化更加明显,甚至可以实现从无条带到有条带的转变。BioTeKe 试剂盒也能实现扩增硅藻DNA 的需要,水样中改良组进行扩增后电泳条带亮度与常规组亮度相似,但组织中改良组进行扩增后电泳条带则更亮。
(1)使用TIANGEN 试剂盒提取水样中硅藻DNA时,常规组可以提取少量DNA并进行PCR扩增,电泳显示较弱条带,但改良组的条带明显变亮。提取组织中硅藻DNA时,常规组提取DNA并进行PCR扩增后的电泳条带不可见,但改良后的电泳条带显示效果较好,与水样硅藻DNA提取的电泳条带亮度接近(图2A)。
(2)使用FOREGENE 试剂盒提取水样中硅藻DNA 时,常规组可以提取少量DNA 并进行PCR 扩增,但扩增产物较少,电泳显示较弱条带,改良后的电泳条带稍亮于常规组的电泳条带。提取组织中硅藻DNA 时,常规组可以提取少量DNA,经过PCR 扩增,电泳显示较弱条带,与常规组水样提取的硅藻DNA 亮度接近,但改良组的条带较改良前明显变亮(图2B)。
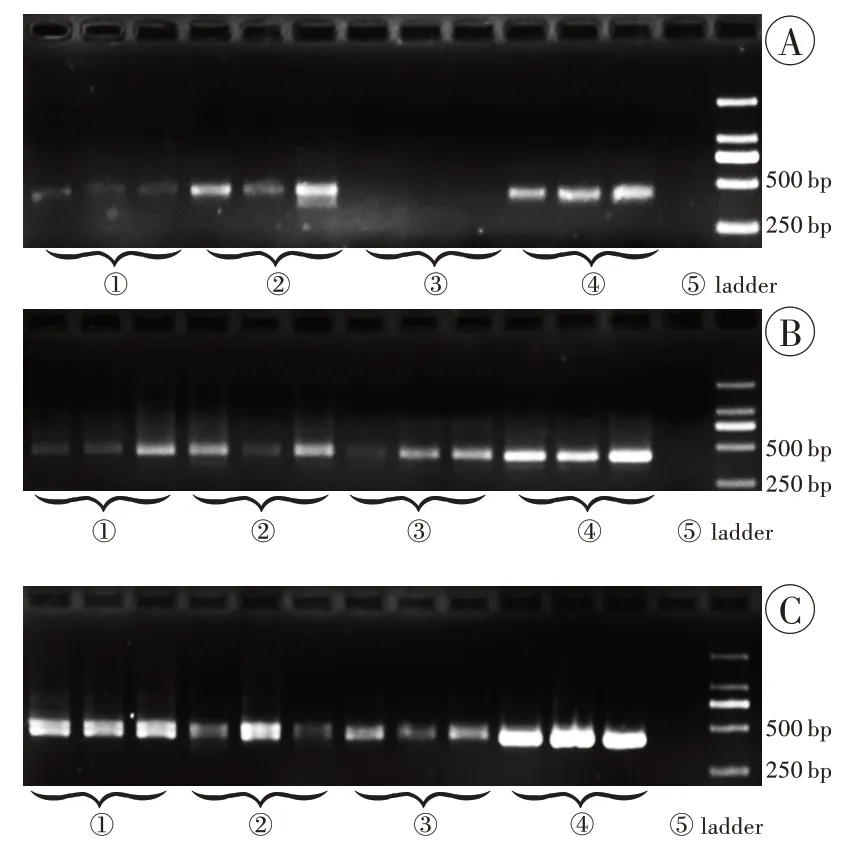
图2 3 种试剂盒提取产物进行PCR 扩增后的电泳结果Fig.2 Electrophoresis results of PCR amplification products extracted from three kits extracts
(3)使用BioTeKe 试剂盒提取水样中硅藻DNA时,常规组与改良组的电泳条带亮度差别不大。而提取组织中硅藻DNA 时,常规组可以提取少量DNA 并进行PCR 扩增,电泳显示稍弱条带,但改良后,电泳显示高亮条带且亮于水样硅藻DNA提取效果(图2C)。
(4)将上述3 种试剂盒与已含玻璃珠涡旋振荡的PowerSoil 试剂盒对水样硅藻DNA 提取后的PCR 扩增产物电泳效果进行对比,可以看出,TIANGEN试剂盒及FOREGENE 试剂盒改良后可以达到PowerSoil 试剂盒的电泳条带亮度,而BioTeKe试剂盒在常规组和改良组对水样硅藻DNA提取后PCR扩增产物的电泳效果均可达到PowerSoil试剂盒的电泳条带亮度(图3A)。
(5)将上述3 种试剂盒与已含玻璃珠涡旋振荡的PowerSoil 试剂盒对组织硅藻DNA 提取后的PCR 扩增产物电泳效果进行对比,可以看出,3 种试剂盒常规组电泳条带不明显,但改良组,可达到PowerSoil 试剂盒的电泳条带亮度(图3B)。
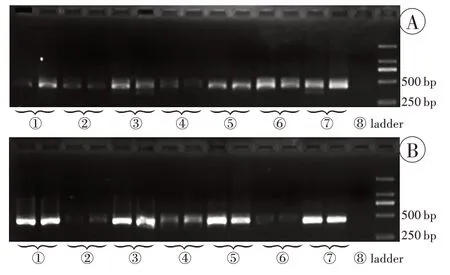
图3 4 种试剂盒提取产物进行PCR 扩增后的电泳结果Fig.3 Electrophoresis results of PCR amplification products extracted from four kits
2.4 qPCR
4种试剂盒对硅藻DNA 提取后分别进行qPCR,获得Ct值,且数据基本稳定。由表2可见:
表2 水样和组织使用3 种试剂盒改良前后获得的Ct值比较Tab.2 Comparison of Ct values of water samples and tissues before and after improvement with three kits(n=5,)
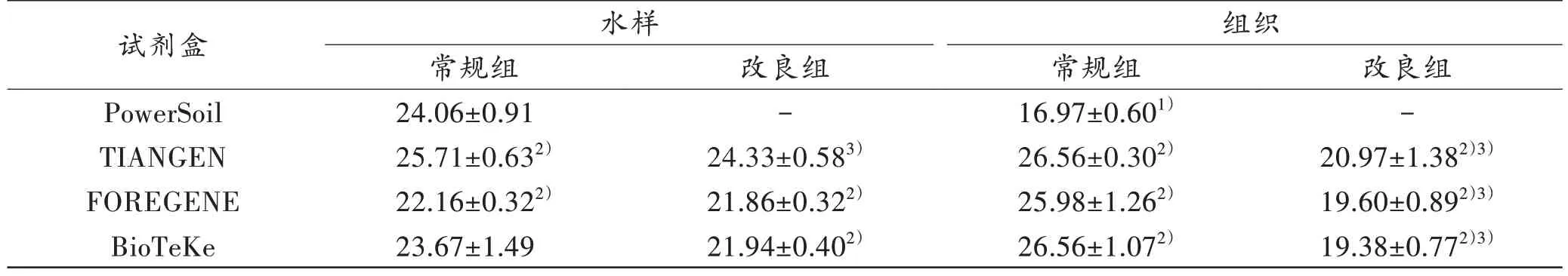
表2 水样和组织使用3 种试剂盒改良前后获得的Ct值比较Tab.2 Comparison of Ct values of water samples and tissues before and after improvement with three kits(n=5,)
注:1)与相同试剂盒水样相比,P<0.05;2)与PowerSoil试剂盒常规组比较,P<0.05;3)与相同试剂盒常规组比较,P<0.05。“-”表示无数据。
(1)PowerSoil试剂盒的水样Ct值大于组织的Ct值(P<0.05)。
(2)TIANGEN 试剂盒常规组水样及组织的Ct值均高于PowerSoil 试剂盒(P<0.05);改良后水样Ct值下降(P<0.05),并与PowerSoil试剂盒接近,改良后组织Ct值下降(P<0.05),但仍高于PowerSoil试剂盒(P<0.05)。
(3)FOREGENE 试剂盒常规组的水样Ct 值低于PowerSoil 试剂盒(P<0.05),改良组水样Ct 值与常规组接近,但仍低于PowerSoil 试剂盒(P<0.05);常规组组织的Ct 值则高于PowerSoil 试剂盒(P<0.05),改良后组织Ct 值下降(P<0.05),但仍高于PowerSoil 试剂盒(P<0.05)。
(4)BioTeKe 试剂盒常规组的水样Ct 值接近于PowerSoil 试剂盒(P>0.05),改良组水样Ct 值变化不明显,但仍低于PowerSoil 试剂盒(P<0.05);常规组组织的Ct 值高于PowerSoil 试剂盒(P<0.05),改良组组织Ct 值较常规组下降(P<0.05),但仍高于PowerSoil试剂盒(P<0.05)。
3 讨论
在硅藻相关领域的研究中,由于仅涉及水样和底栖硅藻样本的检验,所以硅藻DNA 的提取都以植物DNA 提取商品化试剂盒及泥土DNA 提取商品化试剂盒为主要方法,而在法医学实践中,基于从溺死者肺组织提取硅藻DNA 的特殊实践需求,目前国内外均采用土壤DNA 提取商品化试剂盒,其中Power Soil 系列土壤DNA 提取试剂盒应用最多,基本成为法医硅藻DNA 提取的标准试剂盒[10,13-14]。因此,本研究以该试剂盒作为参照标准,检验玻璃珠-涡旋振荡改良方法对法医学硅藻DNA 提取的效果影响。
3.1 玻璃珠-涡旋振荡改良的依据及条件设置
硅藻因坚硬的硅质细胞壁保护而不易破坏,使得普通试剂盒难以提取硅藻DNA,PowerSoil 试剂盒采用磁珠物理击打的方法先破坏硅质外壳,在采用其他方法对DNA 进行纯化提取即可达到更好的效果。因此,在其他试剂盒提取DNA 时,若在消化过程中加入玻璃珠涡旋振荡对硅藻进行有效物理击打,使硅藻DNA 充分释放,则使普通DNA 提取试剂盒也可以对硅藻DNA 更充分地提取。在法医学实践中,对组织中的硅藻DNA 进行提取难度更大,使用玻璃珠击打同样可以使硅藻从组织中充分游离。
DNA 提取中,玻璃珠的直径、振荡强度和时间直接影响DNA 提取的质和量。玻璃珠的大小和直径对实验结果产生重要影响,大的玻璃珠在涡旋振荡过程中对硅藻细胞击打力度较大,直径小的玻璃珠可更充分间隔硅藻细胞,起到分散硅藻的作用,两者通过使用不同质量比的玻璃珠进行混合才可以达到最佳破碎效果。若玻璃珠直径太大碰撞力量大,容易造成DNA 的断裂,不利于硅藻在体系内均匀分散;而直径太小,虽有利于促进硅藻在体系内的均匀分散,但撞击力量减小,不利于硅藻充分破壳。因此,本研究中,选用大小不同、直径分别为2 mm 与0.5 mm 的玻璃珠,采用不同质量比混合,探索使硅壳破碎的最佳比例。针对玻璃珠涡旋振荡时间的确定,在预实验过程中,分别使用5、10、15 min 进行涡旋振荡,发现时间越长、PCR 扩增产物电泳条带越暗,因为DNA 过度涡旋振荡会导致DNA 断裂,使PCR 扩增效果变差[20],因此,本研究在2~6 min 内进行多次重复实验,以缩小最适时间范围。同时对大小玻璃珠质量比和涡旋振荡时间进行控制,电泳结果显示,质量比为1∶1 时涡旋振荡4 min 即可达到最佳涡旋振荡效果,而涡旋振荡时间过长则延长实验时间,同时也会加速实验仪器的损耗。
3.2 改良组植物DNA 提取试剂盒用于硅藻DNA 提取的可行性
TIANGEN 试剂盒DNA 的提取采用非蛋白酶法实现对植物DNA 的提取,改良组水样硅藻DNA 提取特异性扩增条带亮度变化较小,Ct 值显示有差异,改良组DNA 提取量略有提升;而按试剂盒操作对于组织中的硅藻DNA 更是难以提取,特异性扩增后未显条带,应用改良法特异性扩增后条带由无到有,Ct 值降低且差异具有统计学意义(P<0.05)。改良组TIANGEN试剂盒对组织的硅藻DNA 提取量明显提升,可以满足法医学硅藻DNA 分析的需要。而FOREGENE 试剂盒用蛋白酶对植物DNA 进行提取,对水样或者组织均可以进行消化,改良组水样中硅藻DNA 提取Ct 值差异无统计学意义,DNA 含量未见明显提升,组织样本中的硅藻DNA 提取条带显著变亮,Ct 值差异具有统计学意义(P<0.05)。以上研究结果提示,和常规组比较,改良组用于植物提取的2 个试剂盒(TIANGEN试剂盒和FOREGENE 试剂盒)可以提高水样中硅藻DNA 的提取量,对于组织样本改良效果更加显著,甚至可以实现从不能提取到实现高质量提取的转变,普通植物DNA 提取试剂盒亦可以用于法医学硅藻分子检验。
本研究仅应用qPCR 扩增后的Ct 值进行比较,有明显的对比性且与电泳结果相同,若要准确地对DNA 进行定量则应建立标准曲线以获得DNA 含量[21],但标准曲线的建立通常是对纯品定量更为准确,而样本检验中的硅藻为混合物,能否准确定量还有待进一步研究。
3.3 血液DNA 提取试剂盒用于硅藻检验的可行性
由于血液DNA 提取试剂盒是目前使用最广泛、最成熟且最易获得的试剂盒,如果血液DNA 提取试剂盒也能满足硅藻DNA 的提取,将极大便利法医学硅藻检验。
本研究选用了在实验中常见且经济的基于PK 消化法的BioTeKe 试剂盒,发现该试剂盒可以实现硅藻DNA 的提取,在对水样中硅藻进行提取时,无论是否加入玻璃珠均可获得较亮的特异性电泳条带,达到分子检验要求;对组织中硅藻进行DNA 提取也能获得成功,经改良法后,特异性电泳条带显著变亮,Ct值下降(P<0.05),即DNA 提取量显著提高。究其原因,可能是试剂盒中PK[22]使膜蛋白降解,与DNA 结合的蛋白质解离,使DNA 充分游离,硅藻虽然具有坚硬的硅质外壳,但从扫描电子显微镜可知其表面具有大量孔径与细胞内腔相通,因此,PK 可以通过硅藻外壳进入细胞消化细胞结构,使得硅藻内的DNA 释放,同时由于硅藻DNA 的基因组较小,如三角褐指藻基因组大小为27.4 Mb、假微型海链藻基因组大小为34 Mb、阳光管硅藻的基因组大小为49.7 Mb,远小于人类和动物的基因组[23],因此,被PK 消化后释放游离的硅藻DNA 分子极易溢出硅藻细胞,也提升了DNA 提取的效果[22]。本研究中非蛋白酶法进行提取的TIANGEN试剂盒对于组织中硅藻DNA 的提取效果差,即便是加入玻璃珠后,提取效果与基于酶消化法的FOREGENE 试剂盒仍存在一定差距,但由于商家技术保护原因,该试剂盒使用的蛋白酶成分尚不清楚。
硅藻种类繁多,不同种类的硅藻在基因组大小、硅壳上的孔径大小差异很大,基于PK 消化法的DNA提取技术能否实现样品中所有硅藻种类DNA 的提取,尚需后续进一步研究。同时,基于其他原理或者同样基于PK 消化法的其他商品化血液DNA 提取试剂盒是否可用于硅藻检验,在使用前尚需进行逐个检验。但是,由于样品中硅藻含量极低,本研究无法通过琼脂糖电泳对提取产物的DNA 片段大小进行检测,故只能借用PCR 技术的强大倍增功能,选用了硅藻18S rDNA 上的硅藻特异性引物D512、D978 作为检验硅藻DNA 是否提取成功的参考标准,该对引物的扩增产物长度为390~410 bp,该实验条件对更大片段的硅藻DNA 检验是否有影响,仍需进一步研究。已有研究[24-25]表明,不同引物可扩增的硅藻种类不同,因此后续研究也将使用不同区域、不同扩增长度的引物探究对提取效果的影响,同时应用通用引物结合高通量测序来确证硅藻扩增的同一性。
3.4 小结
本研究建立的基于玻璃珠-涡旋振荡改良技术显著提升了植物DNA 提取试剂盒、基于PK 的血液DNA提取试剂盒提取硅藻DNA 的效果,扩大了法医学硅藻DNA 检验试剂盒的选择范围。但本研究样本量不大,在使用改良组方法时,建议进行方法验证和确认。
猜你喜欢
大自然探索(2023年7期)2023-11-14 13:07:36
课堂内外(小学版)(2023年9期)2023-10-11 14:39:26
天天爱科学(2022年9期)2022-09-15 01:12:26
数学大王·中高年级(2022年9期)2022-05-30 10:48:04
小读者·爱读写(2021年11期)2021-12-05 13:44:02
娃娃乐园·综合智能(2021年3期)2021-04-13 02:00:04
科技视界(2020年26期)2020-09-24 03:25:06
科技视界(2020年17期)2020-07-30 14:03:27
黑龙江水利科技(2020年8期)2020-01-12 06:26:00
上海建材(2018年1期)2018-04-18 12:15:16
