
基于TLR4信号通路研究小檗碱对血管紧张素Ⅱ诱导ANA-1细胞炎性应答的影响
2022-06-20 05:44高德兴贾沛芝
康复学报 2022年2期
高德兴,贾沛芝,彭 军,林 炜
福建中医药大学中西医结合研究院,福建 福州 350122
炎性应答是一个错综复杂的生物过程,炎性应答的启动是应对不同刺激的反应[1-2],是人体对微生物感染、免疫细胞浸润、化学刺激以及组织损伤最有效的保护机制之一[3]。持续的损伤或刺激会导致炎性应答的不可控制,从而诱导全身性炎症,并促进慢性炎症的发展,导致器官损伤[4]。模式识别受体Toll样受体4(Toll-like receptor 4,TLR4)作为上游信号分子,与炎性应答以及相关疾病的发生发展过程高度相关[5],TLR4的激活能够启动炎性应答靶基因的转录,产生炎性应答[6-7]。有研究表明,血管紧张素Ⅱ(angiotensinⅡ,AngⅡ)具有明显的促炎效应,参与了炎性应答过程中的关键环节,它能够造成损伤相关分子模式(damage-associated molecular pattern,DAMPs)和新抗原形成,固有免疫细胞被DAMPs激活并诱发固有免疫应答,AngⅡ能够被其中的TLR4识别,并触发机体免疫系统防御反应[8-9]。小檗碱(berberine,BBR)又名黄连素,是从黄连中分离提取的一种天然生物碱,是黄连的主要药物活性物质,它由连接2个甲氧基的萘环和连接双醚基环的苯相接而成,拥有较大的疏水层,能通过与髓样分化蛋白-2(myeloid differentiation protein-2,MD-2)竞争性结合而阻断脂多糖(lipopolysaccharides,LPS)/TLR4复合体的形成,抑制TLR4信号介导的炎性应答,是TLR4信号通路的天然阻断剂,常被用于治疗肠道感染、眼部感染、炎症性肠病等炎症性疾病。AngⅡ诱导的炎性应答可能是通过门户蛋白TLR4激活网络调控下游级联反应,其中包括核因子κB(nuclear factor kappa-B,NF-κB)和丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)的激活以及氧化应激损伤。因此,本课题组提出假说:BBR能够通过抑制TLR4信号通路的激活进而调控AngⅡ诱导的炎性应答。为了进一步探讨其相关的作用机制,本研究使用AngⅡ刺激巨噬细胞(ANA-1)诱导建立体外炎症模型,验证AngⅡ是否能够诱导TLR4及下游信号通路的转导,并基于TLR4信号通路分析BBR对AngⅡ诱导炎性应答的影响。
1 实验材料
1.1 主要药物和细胞株
小檗碱(上海源叶科技有限公司);AngⅡ粉末(美国APExBIO生物科技有限公司);ANA-1细胞株(北京北纳创联生物技术研究院)。
1.2 主要试剂
RPMI-1640(Roswell Park Memorial Institute-1640)培养基(美国Gibco公司);澳洲胎牛血清(美国Gibco公司);青霉素/链霉素混合液(美国Hyclone公司);CCK-8细胞增殖与细胞毒性检测试剂盒(美国APExBIO生物科技有限公司);RIPA裂解液、SDS-PAGE蛋白上样缓冲液(南京碧云天生物技术有限公司,规格5×);Phosphatase抑制剂Cocktail I(美国Abcam公司);PhosStop磷酸酶抑制剂(德国罗氏诊断有限公司);BCA蛋白浓度测定试剂盒(美国Thermo Fisher Scientific公司);快速凝胶制备试剂盒(上海雅酶生物科技有限公司);TLR4抗体、JNK抗体、p38抗体、GAPDH抗体(武汉三鹰生物技术有限公司);MyD88抗体、Phospho-NF-κBp65(Ser536)抗体、NF-κB p65抗体、Phospho-JNK抗体、Phospho-ERK1/2抗体、ERK1/2抗体、Phospho-p38抗体、p38抗体、IRF3抗体、Phospho-IRF3抗体(美国Cell Signaling Technology公司);TLR4抑制剂(TAK-242)(美国MedChemExpress公司);二甲基亚砜(Dimethyl sulfoxide,DMSO)溶液(北京索莱宝科技有限公司);磷酸盐缓冲溶液(phosphate buffer saline,PBS)(美国Hyclone公司)。
1.3 主要仪器
37℃CO2培养箱(美国Thermo Forma公司);超净工作台(苏州净化设备公司);倒置显微镜系统(德国Leica仪器有限公司);电泳仪、电泳槽、小型转膜仪、化学发光成像系统(美国Bio-Rad公司);连续波长多功能酶标仪(奥地利TECAN公司)。
2 实验方法
2.1 细胞分组与培养
2.1.1 细胞分组 BBR粉末溶解于DMSO溶液中,配制成20 mmol/L母液,按需用无血清1640培养基稀释成浓度为1、5、10μmol/L的工作液,并避光分装放置于-20℃冰箱保存。AngⅡ粉末溶解于PBS溶液,配制成1 mmol/L母液,按需用无血清1640培养基稀释成浓度为1μmol/L的工作液,分装放置于-80℃冰箱保存。ANA-1细胞株培养于RPMI-1640完全培养基中,置于37℃、5%CO2、饱和湿度的细胞培养箱内。将对数增长期的ANA-1细胞按照随机数字表法平均分为对照组、荧光抗体组、模型组、低剂量BBR组、中剂量BBR组、高剂量BBR组、抑制剂组。
2.1.2 细胞培养 ①对照组:皿中加入2.5μL的DMSO;②荧光抗体组:皿中加入2.5μL的DMSO;③模型组:皿中加入浓度为1μmol/L的AngⅡ工作液;④低剂量BBR组:皿中加入浓度为1μmol/L的BBR工作液;⑤中剂量BBR组:皿中加入浓度为5μmol/L的BBR工作液;⑥高剂量BBR组:皿中加入浓度为10μmol/L的BBR工作液;⑦抑制剂组:皿中加入浓度为10μmol/L的TAK242工作液。
2.2 观察指标
2.2.1 细胞增殖与毒性检测实验方法检测细胞活力 取对数生长期ANA-1细胞以1×105个/mL的密度接种于96孔板培养24 h后,弃培养基,低、中、高剂量BBR组分别加入1、5、10μmol/L工作液100μL干预24 h后,弃培养基,加入CCK-8溶液培养2 h后,在450 nm波长下测吸光度(Absorbance,Abs)值(A值)。每个浓度至少做3个复孔。采用细胞增殖与毒性检测实验方法(cell counting kit-8,CCK-8)检测对照组,低、中、高剂量BBR组的细胞A值。
2.2.2 流式细胞术检测细胞膜上TLR4平均荧光强度 取对数生长期ANA-1细胞按2.5×105个/mL的密度接种于35 mm培养皿中,待细胞生长至汇合度达60%~70%,弃原培养基,改用无血清1640培养基培养6 h后,模型组用1μmol/L AngⅡ刺激15 min;模型+小檗碱高剂量组用高剂量BBR预处理2 h后,用1μmol/L AngⅡ刺激15 min,弃上清,用PBS溶液将细胞吹打下来,制备成单细胞悬液,离心后弃上清。荧光抗体组、模型组、模型+小檗碱高剂量组均分别加入TLR4荧光抗体(预冷1×PBS配制)100μL,避光孵育15~30 min;之后于1 000 r/min离心机上离心3 min;最后用预冷PBS清洗细胞3次后,采用流式细胞术检测TLR4的平均荧光强度(The mean fluorescence intensity,MFI)。
2.2.3 ELISA法检测白细胞介素-1β、白细胞介素-6、肿瘤坏死因子-α的含量 取对数生长期ANA-1细胞以2.5×105个/mL的密度接种于60 mm培养皿中,弃原培养基,改用无血清1640培养基培养6 h后,模型组加入1μmol/L AngⅡ处理细胞24 h;模型+低剂量BBR组、模型+中剂量BBR组、模型+高剂量BBR组、模型+抑制剂组加入不同浓度BBR或TAK 242预处理2 h后,用1μmol/L AngⅡ处理细胞24 h,收集培养基上清液备用。分别按顺序加入标准品和各组样本,作复孔。加酶联亲和物并均匀振荡后置于37℃恒温中1 h,然后弃上清,清洗后加入底物,避光保存15 min。在各孔中加入反应终止液。采用ELISA法检测白细胞介素-1β(interleukin-1β,IL-1β)、白细胞介素-6(interleukin-6,IL-6)、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)的含量。
2.2.4 实时定量PCR法检测IL-1β、IL-6、TNF-α mRNA的水平 细胞培养及铺板操作如“2.2.3”项所述,模型组加入1μmol/L AngⅡ处理细胞6 h;模型+低剂量BBR组、模型+中剂量BBR组、模型+高剂量BBR组分别加入1、5、10μmol/L BBR预处理2 h;模型+抑制剂组加入10μmol/L TAK242抑制剂预处理2 h。各组分别用1μmol/L AngⅡ处理细胞6 h,用Trizol法提取RNA,遵照cDNA合成试剂盒进行逆转录并扩增,采用实时定量PCR法(quantitativereal-time PCR,Q-PCR)检测IL-1β、IL-6、TNF-α mRNA的水平。引物序列如表1。
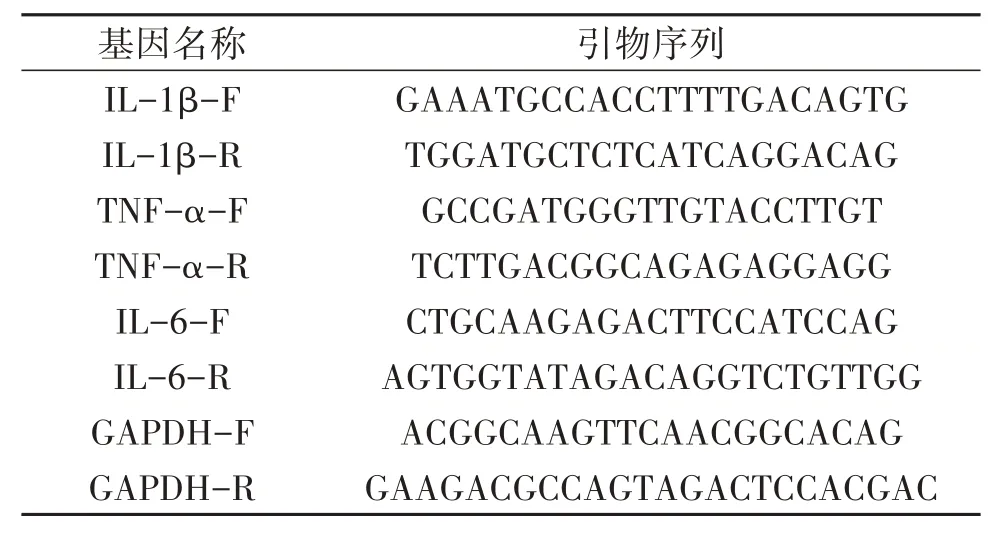
表1 引物序列Table 1 Primer sequence
2.2.5 Western blot法检测TLR4信号通路相关蛋白的表达 模型组加入1μmol/L AngⅡ处理细胞15、30、45 min以及12 h;模型+低剂量BBR组、模型+中剂量BBR组、模型+高剂量BBR组分别加入1、5、10μmol/L BBR预处理2 h;模型+抑制剂组加入10μmol/L TAK242抑制剂预处理2 h。各组分别用1μmol/L AngⅡ刺激15、30、45 min以及12 h,收集细胞用于Western blot检测。将细胞用RIPA裂解液提取总蛋白,蛋白质定量法(bicinchoninic acid,BCA)测定蛋白浓度。各蛋白样品均加入1/4总蛋白体积的十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDSPAGE)蛋白上样缓冲液,在100℃金属锅中变性5~10 min后,使用SDS-PAGE电泳法进行凝胶电泳,每个孔上样量30μL,电泳条件:80 V 30 min、100 V 75 min,电泳结束后将蛋白转移到0.22μm聚偏氟乙烯(polyvinylidene fluoride,PVDF)膜,转膜条件:100 V 90 min。转膜结束后,用5%脱脂奶粉封闭1~2 h后加入相应一抗4℃孵育过夜。采用辣根过氧化物酶标记的二抗(1∶5 000)常温孵育1~2 h后,用Bio-Rad凝胶成像系统进行图像分析。采用Western blot检测p-p65、p65、p-IRF3、IRF3、p-ERK、ERK、p-p38、p38、JNK、p-JNK、TLR4、MyD88、p-IκBα、IκBα、AP-1蛋白的表达。
2.2.6 生化法检测丙二醛含量和超氧化物歧化酶活力 模型组加入1μmol/L AngⅡ处理细胞12 h;模型+低剂量BBR组、模型+中剂量BBR组、模型+高剂量BBR组分别加入1、5、10μmol/L BBR预处理2 h。各组分别用1μmol/L AngⅡ刺激15、30、45 min以及12 h,收集细胞上清用于检测丙二醛(malondialdehyde,MDA)和超氧化物歧化酶(superoxide dismutase,SOD)。按照MDA及SOD测定试剂盒说明书配制好上清混合液。将200μL上清液混合液分别加进96孔板,做好标记,使用酶标仪检测A值,波长为532 nm,按照说明书进行MDA含量和SOD活力计算。
2.3 统计学方法
采用SPSS24.0软件进行数据分析。计量资料服从正态分布,数据采用(±s)表示,多组间比较采用单因素方差分析,两两比较采用Bonferroni检验。P<0.05为差异具有统计学意义。
3 结 果
3.1 4组ANA-1细胞活力比较
与对照组比较,低、中、高剂量BBR组细胞活性均无明显区别,差异无统计学意义(P>0.05)。见表2。
3.2 4组TLR4 MFI比较
与荧光抗体组比较,模型组MFI明显减弱;与模型组比较,模型+高剂量BBR组MFI明显增强,差异具有统计学意义(P<0.05)。见表3。
表2 4组ANA-1细胞活力比较(±s)Table 2 Comparison of the activity of ANA-1 cells in four groups(±s)

表2 4组ANA-1细胞活力比较(±s)Table 2 Comparison of the activity of ANA-1 cells in four groups(±s)
组别对照组低剂量BBR组中剂量BBR组高剂量BBR组n3 3 3 3细胞活性(吸光值)0.29±0.04 0.29±0.01 0.29±0.05 0.29±0.06
表3 4组TLR4平均荧光强度比较(±s)Table 3 Comparison of the mean fluorescence intensity of TLR4 in four groups(±s)
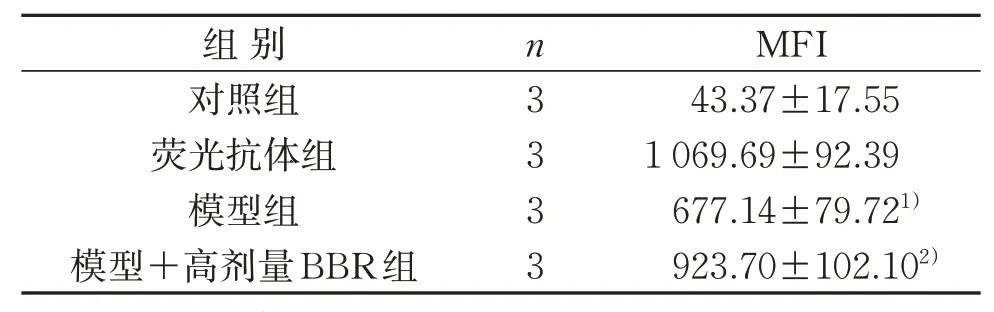
表3 4组TLR4平均荧光强度比较(±s)Table 3 Comparison of the mean fluorescence intensity of TLR4 in four groups(±s)
注:与荧光抗体组比较,1)P<0.05;与模型组比较,2)P<0.05。Note:Compared with the fluorescent antibody group,1)P<0.05;compared with the model group,2)P<0.05.
组别对照组荧光抗体组模型组模型+高剂量BBR组n3 3 3 3 MFI 43.37±17.55 1 069.69±92.39 677.14±79.721)923.70±102.102)
3.3 6组IL-1β、IL-6、TNF-αmRNA水平比较
与对照组比较,模型组IL-1β、IL-6、TNF-α mRNA水平明显升高;与模型组比较,模型+低、中、高剂量BBR组IL-1β、IL-6、TNF-αmRNA水平均明显降低,差异具有统计学意义(P<0.05)。见表4。此外,研究结果显示,与模型组比较,模型+抑制剂组、模型+高剂量BBR组IL-1β、IL-6、TNF-αmRNA水平均明显降低,差异具有统计学意义(P<0.05)。见表5。
表4 6组IL-1β、IL-6、TNF-αmRNA水平的比较(±s)Table 4 Comparison of mRNA levels of IL-1β,IL-6 and TNF-αin six groups(±s)

表4 6组IL-1β、IL-6、TNF-αmRNA水平的比较(±s)Table 4 Comparison of mRNA levels of IL-1β,IL-6 and TNF-αin six groups(±s)
注:与对照组比较,1)P<0.05;与模型组比较,2)P<0.05。Note:Compared with the control group,1)P<0.05;compared with the model group,2)P<0.05.
组别对照组模型组高剂量BBR组模型+低剂量BBR组模型+中剂量BBR组模型+高剂量BBR组n3 3 3 3 3 3 IL-1βmRNA 1.00±0.20 1.52±0.191)0.95±0.28 0.99±0.372)0.54±0.132)0.37±0.092)IL-6 mRNA 1.00±0.07 4.78±0.201)1.03±0.12 0.86±0.212)0.85±0.112)0.75±0.082)TNF-αmRNA 1.00±0.12 1.85±0.261)1.05±0.23 0.80±0.312)0.70±0.192)0.61±0.042)
表5 6组IL-1β、IL-6、TNF-αmRNA水平比较(±s)Table 5 Comparison of mRNA levels of IL-1β,IL-6 and TNF-αin six groups(±s)

表5 6组IL-1β、IL-6、TNF-αmRNA水平比较(±s)Table 5 Comparison of mRNA levels of IL-1β,IL-6 and TNF-αin six groups(±s)
注:与对照组比较,1)P<0.05;与模型组比较,2)P<0.05。Note:Compared with the control group,1)P<0.05;compared with the model group,2)P<0.05.
组别对照组模型组抑制剂组模型+抑制剂组高剂量BBR组模型+高剂量BBR组n3 3 3 3 3 3 IL-1βmRNA 1.00±0.05 2.54±0.181)1.06±0.15 0.64±0.142)0.83±0.24 0.19±0.022)IL-6 mRNA 1.00±0.19 2.16±0.261)0.83±0.13 0.85±0.242)0.93±0.09 0.43±0.122)TNF-αmRNA 1.00±0.07 1.61±0.161)1.00±0.17 0.40±0.052)0.81±0.11 0.23±0.042)
3.4 6组IL-1β、IL-6、TNF-α含量比较
与对照组比较,模型组IL-1β、IL-6、TNF-α的含量明显升高;与模型组比较,模型+低、中、高剂量BBR组IL-1β、IL-6、TNF-α含量均明显降低,差异具有统计学意义(P<0.05)。见表6。此外,研究结果显示,与模型组比较,模型+高剂量BBR组及模型+抑制剂组IL-1β、IL-6、TNF-α含量均明显降低,差异具有统计学意义(P<0.05)。见表7。
表6 6组IL-1β、IL-6、TNF-α含量比较(±s) pg/mLTable 6 Comparison of content of IL-1β,IL-6 and TNF-αin six groups(±s) pg/mL

表6 6组IL-1β、IL-6、TNF-α含量比较(±s) pg/mLTable 6 Comparison of content of IL-1β,IL-6 and TNF-αin six groups(±s) pg/mL
注:与对照组比较,1)P<0.05;与模型组比较,2)P<0.05。Note:Compared with the control group,1)P<0.05;compared with the model group,2)P<0.05.
组别对照组模型组高剂量BBR组模型+低剂量BBR组模型+中剂量BBR组模型+高剂量BBR组n3 3 3 3 3 3 IL-1β 5.79±0.27 6.98±0.021)5.56±0.10 6.61±0.072)6.13±0.112)4.10±0.292)IL-6 13.63±0.21 32.65±1.061)13.26±0.69 10.62±0.292)4.13±0.282)3.33±0.132)TNF-α 105.48±0.79 429.34±7.791)105.96±0.62 111.13±0.372)90.99±0.792)73.67±0.992)
表7 6组IL-1β、IL-6、TNF-α含量比较(±s) pg/mLTable 7 Comparison of content of IL-1β,IL-6 and TNF-αin six groups(±s) pg/mL

表7 6组IL-1β、IL-6、TNF-α含量比较(±s) pg/mLTable 7 Comparison of content of IL-1β,IL-6 and TNF-αin six groups(±s) pg/mL
注:与对照组比较,1)P<0.05;与模型组比较,2)P<0.05。Note:Compared with the control group,1)P<0.05;compared with the model group,2)P<0.05.
组别对照组模型组抑制剂组模型+抑制剂组高剂量BBR组模型+高剂量BBR组n3 3 3 3 3 3 IL-1β 5.36±0.21 6.66±0.071)4.52±0.37 4.36±0.322)5.39±0.24 5.14±0.032)IL-6 10.41±0.31 35.50±0.351)6.32±2.27 5.62±0.422)11.55±0.09 10.28±0.372)TNF-α 101.87±0.92 434.96±14.071)86.18±1.05 73.67±1.652)98.96±0.84 93.32±2.022)
3.5 6组AngⅡ干预后TLR4相关蛋白磷酸化表达比较
3.5.1 6组AngⅡ干预45 min TLR4相关蛋白磷酸化表达比较 与对照组比较,模型组干预45 min后p65、IRF-3蛋白的磷酸化表达明显上调,模型+低、中、高剂量BBR组干预45 min后p65、IRF-3蛋白的磷酸化表达明显下调。见图1。
3.5.2 6组AngⅡ干预15、30、45 min TLR4相关蛋白磷酸化表达比较 与对照组比较,模型组干预15 min后ERK、p38蛋白磷酸化表达明显上调,干预30 min后JNK蛋白磷酸化表达明显上调,干预45 min后p65、IRF-3蛋白磷酸化表达明显上调。而模型+高剂量BBR组、模型+抑制剂组p65、IRF-3、ERK、p38、JNK蛋白磷酸化表达均明显下调。见图2。
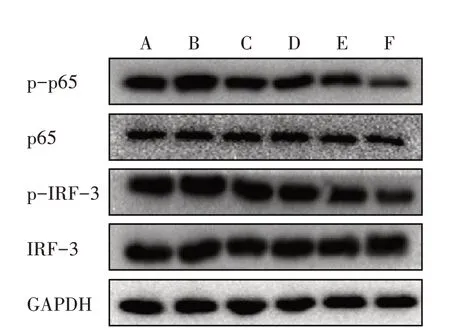
图1 6组干预45 min TLR4相关蛋白磷酸化表达比较Figure 1 Comparison of phosphorylation expression of TLR4-related protein after intervention for 45 min in six groups
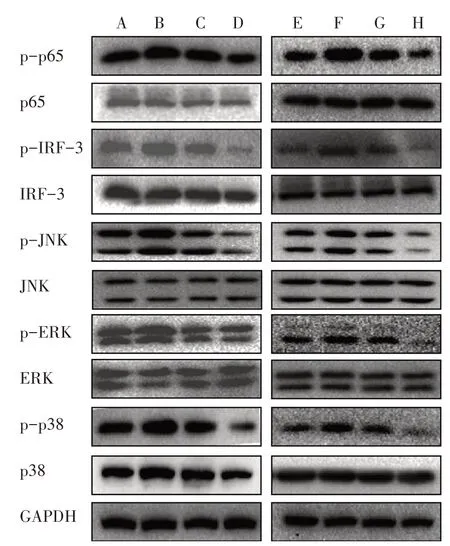
图2 6组AngⅡ干预15、30、45 min TLR4相关蛋白磷酸化的表达比较Figure 2 Comparison of phosphorylation expression of TLR4-related protein after intervention for 15,30,45 min in six groups
3.6 6组AngⅡ干预后TLR4相关蛋白磷酸化表达比较
3.6.1 6组AngⅡ干预12 h TLR4相关蛋白磷酸化表达比较 与对照组比较,模型组干预12 h后TLR4、MyD88、AP-1蛋白的表达以及p65、IκBα、JNK蛋白的磷酸化表达明显上调。模型+低、中、高剂量BBR组TLR4、MyD88、AP-1蛋白的表达以及p65、IκBα、JNK蛋白的磷酸化表达均明显下调。见图3。
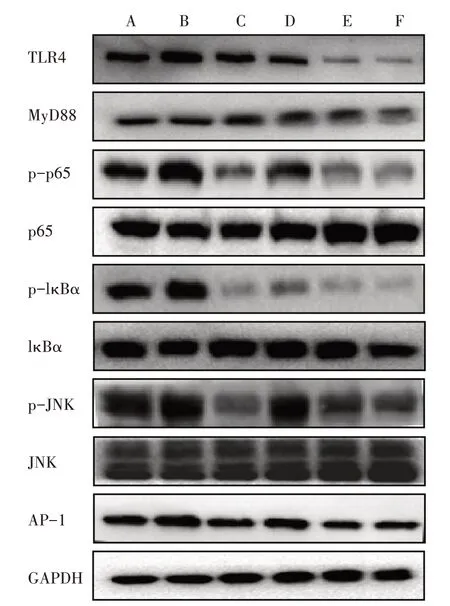
图3 6组干预12 h TLR4相关蛋白磷酸化表达比较Figure 3 Comparison of phosphorylation expression of TLR4-related protein after intervention for 12 h in six groups
3.6.2 5组AngⅡ干预12 h TLR4相关蛋白磷酸化表达比较 与对照组比较,模型组干预12 h后TLR4、p-p65的表达明显上调,而模型+高剂量BBR组、模型+抑制剂组、模型+抑制剂+高剂量BBR组TLR4、p-p65蛋白表达均明显下调。见图4。
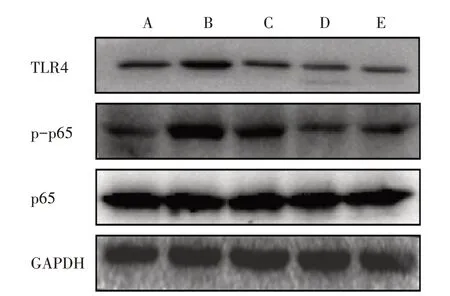
图4 5组干预12 h TLR4相关蛋白磷酸化表达比较Figure 4 Comparison of phosphorylation expression of TLR4-related protein after intervention for 12 h in five groups
3.7 5组MDA含量和SOD活力比较
与对照组比较,模型组MDA含量明显上调,SOD活力明显下调,差异具有统计学意义(P<0.05)。模型+低、中、高剂量BBR组MDA含量均明显下调,SOD活力明显上调,差异具有统计学意义(P<0.05)。见表8。
表8 5组MDA含量和SOD活力比较(±s) nmol/mLTable 8 Comparison of MDA content and SOD activity in five groups(±s) nmol/mL
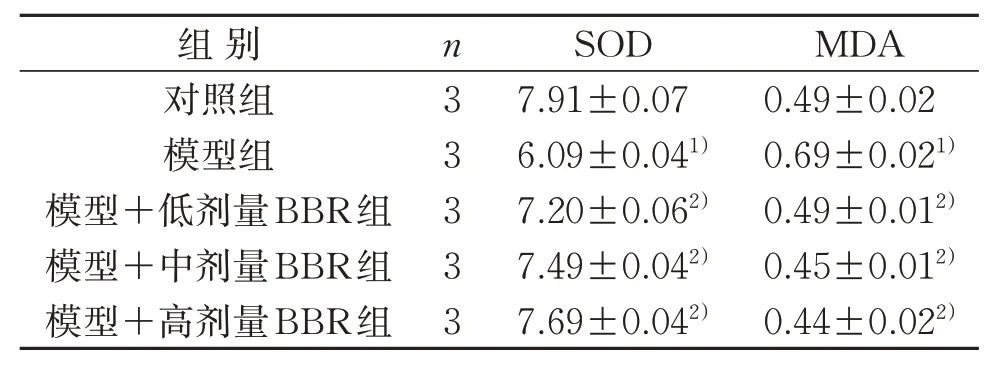
表8 5组MDA含量和SOD活力比较(±s) nmol/mLTable 8 Comparison of MDA content and SOD activity in five groups(±s) nmol/mL
注:与对照组比较,1)P<0.05;与模型组比较,2)P<0.05。Note:Compared with the control group,1)P<0.05;compared with the model group,2)P<0.05.
组别对照组模型组模型+低剂量BBR组模型+中剂量BBR组模型+高剂量BBR组n3 3 3 3 3 SOD 7.91±0.07 6.09±0.041)7.20±0.062)7.49±0.042)7.69±0.042)MDA 0.49±0.02 0.69±0.021)0.49±0.012)0.45±0.012)0.44±0.022)
4 讨 论
4.1 BBR能有效抑制TLR4蛋白“内吞”
由于配体的刺激作用,活化的TLR4分别通过招募MyD88接头蛋白TRIF、TRAM后启动下游信号,同时使配体与TLR4复合体转移至胞内,细胞膜表面的TLR4受体数量减少[10-11]。本研究结果显示,与荧光抗体组比较,模型组MFI明显减弱,这提示AngⅡ诱导了细胞膜上TLR4蛋白数量减少,即发生“内吞”,这可能是因为AngⅡ与细胞表面TLR4结合,内吞转移至胞质内。与模型组比较,模型+高剂量BBR组MFI明显增强,这提示BBR能够阻断AngⅡ诱导的TLR4蛋白“内吞”。这可能与以下因素有关:AngⅡ能够与TLR4的辅助蛋白MD2结合进而招募TLR4,这种直接的相互作用类似于LPS与MD2的结合,导致AngⅡ-MD2-TLR4复合物的形成,激活信号转导以及接头蛋白的募集[12],但这一过程并不依赖于AT1R信号通路[13]。BBR可能通过竞争性抑制AngⅡ与MD2的结合来抑制细胞膜表面的TLR4减少,这一作用可能和BBR竞争性抑制LPS与MD2的结合类似。
4.2 BBR能有效抑制TLR4信号通路的转导和IL-1β、IL-6、TNF-α的转录
研究显示,TLR4的胞内区信号主要通过2条途径被激活,MyD88依赖非依赖途径活化下游信号转导[14-15],最终MAPK、NF-κB以及IRF3信号通路被激活,调节炎性应答及促炎因子(如TNF-α、IL-6、IL-1β)的表达,导致炎性应答的发生发展[16-17]。本研究结果显示,AngⅡ诱导ANA-1细胞15、30、45 min能够诱导ERK、p38、JNK、p65、IRF-3的磷酸化,而TAK242抑制剂的干预均能够抑制上述蛋白的磷酸化水平,这提示了AngⅡ瞬时刺激可能会导致ERK、p38、JNK、p65、IRF-3蛋白磷酸化,即使在AngⅡ诱导12 h(滞后性效应)后依然能够促进TLR4、MyD88、IκBα、AP-1的蛋白表达以及JNK的磷酸化。而模型+高剂量BBR组、模型+抑制剂组p65、IRF-3、ERK、p38、JNK蛋白的磷酸化表达均明显下调,这提示BBR能够有效抑制上述蛋白及磷酸化表达。IL-1β、IL-6、TNF-α是炎性应答触发后产生的细胞因子,当巨噬细胞被外界刺激后,会产生大量促炎因子和早期炎性应答标记物,在组织损伤、微生物感染和免疫系统激活的条件下分泌和释放[18-20]。有研究表明,抑制TLR4/MAPK以及NF-κB信号通路可以进而抑制炎症因子IL-1β、IL-6、TNF-α转录[21-22]。本研究结果显示,AngⅡ能够诱导IL-1β、IL-6、TNF-α mRNA水平及含量明显上调,而BBR、TLR4信号通路抑制剂TAK242均能够明显下调AngⅡ诱导的上述促炎因子mRNA水平及含量。这可能与以下因素有关:①AngⅡ能够诱导TLR4的活化,激活下游级联反应,其中包括激活MAPK信号通路关键蛋白ERK、p38、JNK的磷酸化以及AP-1蛋白水平,并且能够使IκBα蛋白与NF-κB解离并磷酸化,激活NFκB信号通路,从而调控了炎症因子(如IL-1β、IL-6、TNF-α等)的转录。②BBR能够通过TLR4信号通路抑制AngⅡ诱导的相关蛋白表达,进而抑制促炎因子IL-1β、IL-6、TNF-α的转录,即BBR能够通过TLR4信号通路抑制AngⅡ诱导的炎性应答。
4.3 BBR能有效抑制氧化应激反应
TLR4信号通路的激活能够刺激还原型辅酶Ⅱ(nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶[23],导致活性氧(reactive oxygen species,ROS)转化形成[24]。ROS水平的失衡激活NF-κB途径,导致NF-κB的磷酸化和核转位,促进IL-1β、IL-6、TNF-α的分泌,增加炎性应答反应程度[25]。本研究结果显示,模型组MDA含量明显上调,SOD活力明显下降,说明AngⅡ能够诱导ANA-1细胞的氧化应激损伤。而模型+低、中、高剂量BBR组MDA含量均明显下调,SOD活力明显上调,这提示,低、中、高剂量BBR能够改善AngⅡ诱导的ANA-1细胞的脂质过氧化程度及抗氧化能力,降低AngⅡ造成的氧化应激损伤,保护ANA-1细胞,进而降低炎性应答的发生。这可能与以下因素有关:BBR通过抑制TLR4信号通路的激活进而抑制氧化应激的发生发展。BBR抑制氧化应激反应,间接抑制了ROS激活NF-κB信号通路的作用,维持活性氧水平和抗氧化能力的动态平衡,在炎性应答中发挥了重要的作用。
5 小 结
BBR能够通过抑制TLR4蛋白的“内吞”,从而抑制AngⅡ诱导的TLR4信号通路的异常激活,并且抑制下游NF-κB和MAPK信号的转导,降低促炎因子IL-1β、IL-6、TNF-αmRNA水平和含量,调控炎性应答。此外,BBR还能够改善AngⅡ诱导的ANA-1细胞的脂质过氧化程度的增加和抗氧化能力的减弱,降低AngⅡ造成的氧化应激损伤,保护ANA-1细胞。AngⅡ诱导炎性应答的发生,造成靶器官损伤,导致心脑血管疾病、胰岛素抵抗、肾脏疾病、肺动脉高压等疾病病理发展过程。其具体作用机制可能是BBR可通过竞争性抑制TLR4信号通路的激活,减轻炎性应答。这也许可以为临床治疗AngⅡ刺激产生的炎症性疾病提供新思路,但需要进一步通过大样本临床随机对照试验加以验证。此外,BBR对TLR4信号通路的抑制作用是否呈剂量依赖性也有待进一步研究。
猜你喜欢
中国比较医学杂志(2022年8期)2022-11-21
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中风与神经疾病杂志(2022年9期)2022-10-19
中国现代医生(2022年21期)2022-08-22
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
医学概论(2022年4期)2022-04-24
中国药房(2022年7期)2022-04-14
波谱学杂志(2022年1期)2022-03-15
科教导刊·电子版(2018年9期)2018-06-07
