
湿法消解-ICP-AES 内标法测定铑派克废料中的铑
2022-06-18 02:36张玉德金云杰马王蕊
贵金属 2022年1期
刘 伟,刘 文,张玉德,金云杰,林 波,罗 仙,马王蕊,崇 彪
(贵研资源(易门)有限公司 贵研铂业股份有限公司,昆明 650106)
铑派克是三苯基膦乙酰丙酮羰基铑(ROPAC)的常用名,是一种主要起加氢醛化、甲酰化和羰基化作用的均相催化剂[1]。采用铑派克催化剂的低压羰基合成工艺具有选择性高、反应快、成本低、条件温和等优点,是目前世界上生产丁醇和辛醇的主要方法。使用后的铑派克催化剂有部分溶解于副反应产生的酯类、烃类等有机物中,成为油状废料,有机物和磷含量高,铑的含量范围为100~1500 g/t。由于铑的资源稀缺、价格昂贵,从各种铑废料中回收提取铑是铑的重要供应链之一。确定油状铑派克废料中铑含量,是铑派克废料交易和回收提纯铑的重要前提条件。因此,准确、快速测定铑派克废料中的铑含量具有重要意义。
对铑的分析要求,随着分析对象的变化、金属含量高低的不同以及共存成分的复杂程度而有所区别。常用的铑含量的测定方法有化学法和仪器法[2],如铑含量>5%的铑化合物分析,要求相对允许差≤0.5%,通常采用重量法(化学方法)[3];铑含量<5%且共存成分较多的复杂废料,要求测定相对允许差≤2%,可采用分光光度法(UV-vis)[4]、原子吸收分光光度法(AAS)[5]、电感耦合等离子体发射光谱法(ICP-AES)[6-9]、电感耦合等离子体质谱法(ICP-MS)[10-11]等仪器分析方法。
ICP-AES 法具有多元素快速测定、线性范围宽、检出限低等优点,在铂族金属分析中的应用较AAS法和紫外可见分光光度法更普遍、更快捷。但是由于铑的价格昂贵,在废料交易中对铑的分析结果准确度和精密度要求较高,而ICP-AES 仪器固有的波动往往在2%左右,再叠加其它不确定因素,测定结果波动甚至更高,因此改善ICP-AES 测定的稳定性成为测定铑急需解决的重要问题。传统的原子发射光谱法采用同时测定主元素内标线控制波动,采用外加元素做内标的ICP-AES 法也有报道,文献[12]采用钇作内标元素ICP-AES 测定碘化铑中20%的铑,相对标准偏差RSD(n=5)<1%,较不用内标的ICP-AES 法精密度明显改善,准确度接近测定铑的经典方法硝酸六氨合钴重量法。
本文针对铑派克废料,为提高检测速度,保证测定精密度、再现性和准确度,采用硝酸+硫酸+高氯酸冒烟消解样品,在试液和校准曲线溶液中加入定量的钇,用ICP-AES 内标法测定铑含量方法准确、快速,已应用于生产分析。
1 实验
1.1 试剂和样品
所用试剂盐酸、硝酸、硫酸、高氯酸等均为分析纯试剂;铑标准贮存溶液(1000 μg/mL,10%盐酸介质)购自国家钢铁材料测试中心钢铁研究总院。仪器用氩气纯度大于99.99%。实验用水为去离子水。
铑标准溶液(50.0 µg/mL):移取10.00 mL 铑标准贮存溶液于200 mL 容量瓶中,加入20 mL 盐酸,用水稀释至刻度,混匀。
钇标准溶液(100 µg/mL):称取0.5079 g 三氧化二钇于200 mL 烧杯中,加入15 mL 盐酸,低温溶解完全,冷却。加入90 mL 盐酸,转入1000 mL 容量瓶中,用水稀释至刻度,混匀。
实验用铑派克样品为油状,其中sy-1#样品(管理样)参考值Rh 750±10 g/t),SF03130-1#(管理样)参考值Rh 360±5 g/t,其余样品为生产分析样品。
1.2 实验方法
1.2.1 样品试液制备
油状样品混匀后,称取约0.6 g 试样于300 mL烧杯中,加入3 mL 硝酸,0.5 mL 硫酸,盖上表面皿,低温加热至冒白烟,溶液变黑褐色,加入2 mL硝酸,加热至溶液清亮。取下,冷却。加入2 mL硝酸、1 mL 高氯酸,加热至冒白烟,取下,冷却,补加2 mL 硝酸,加热至冒白烟,溶液清亮后取下,冷却。加入10 mL 盐酸,加热煮沸,取下,冷却。用水洗涤烧杯和表面皿,转入100 mL 容量瓶,准确加入1 mL 钇标准溶液,定容,摇匀。
1.2.2 校准曲线溶液制备
移取一定量的铑标准溶液和钇标准溶液,配制成一组钇的质量浓度均为1.0 mg/L,铑的质量浓度依次为0、0.50、1.00、2.50、5.00 和12.50 mg/L 的铑校准曲线溶液,介质为(1+9)盐酸。
1.2.3 测定
使用美国PE 公司Avio 500 型ICP-AES 进行测定,分析线为Rh 343.489 nm,内标线为Y 371.029 nm。
电感耦合等离子体发射光谱仪运行稳定后,选择射频功率1.3 kW、载气流速0.2 L/min、保护气流速0.7 L/min、冷却气流速12 L/min、进样速率1.0 mL/min、径向观测、预燃时间30 s、自动积分、积分两次取平均值。
在上述选定的条件下,用配制好的校准曲线溶液进行标准化,测定样品试液,仪器根据校准曲线自动进行数据处理并输出铑的质量浓度,再计算得出铑的含量。
2 结果与讨论
2.1 样品处理方法对比
称取铑派克废料sy-1#样品7 份,按实验方法处理后测定铑。另外称取6 份试样做对照,采用传统的火试金富集ICP-AES 法测定铑,结果列于表1 和表2。
根据表1 和表2 的数据,两种样品处理方式获得的结果与参考值吻合,相对标准偏差(RSD)均小于1%。对比两种处理方法,火试金富集法取样量大,需要用铅熔炼,污染大,加入铂、金保护灰吹,成本高,铑富集后金属扣溶解困难,需要消化罐密闭溶解,时间长,操作繁琐,劳动强度大;湿法消解则成本低,简便快速,90 min 即可处理一批次样品。选择称样量为0.6 g,采用湿法消解作为样品处理方法,可以大大降低污染和成本,提高分析速度。基于快速测定、绿色环保和节能降耗的原则,确定采用湿法消解作为样品前处理方法。
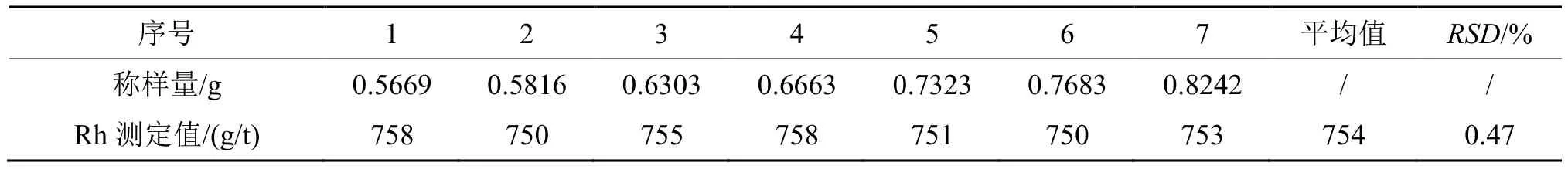
表1 湿法消解实验结果Tab. 1 Experimental results of wet digestion

表2 火试金富集实验结果Tab. 2 Experimental results of fire assaying pre-concentration
2.2 湿法消解条件的选择
称取铑派克废料(sy-1#)样品9 份,分为3 组,使用3 种条件进行湿法消解:1) 150℃,10 mL 硝酸加热1 h,再加1 mL 高氯酸和6 mL 硝酸反复冒烟除去有机物,盐酸溶解残渣。2) 200℃,5 mL 硝酸+0.5 mL 硫酸加热0.5 h,再加1 mL 高氯酸和4 mL硝酸冒烟除去有机物,盐酸溶解残渣。3) 200℃,20 mL 硫酸加热1.5 h,滴加双氧水20 mL,加热1.5 h,盐酸溶解残渣。定容得到的试液按1.2.3 的方法测定,测定结果列于表3。
由表3可见,条件1) 和条件2) 消解效果较好,所得结果与参考值吻合;条件3) 在消解过程中产生大量泡沫,易粘附在烧杯上,消解效果差,所得结果偏低。条件1) 使用的硝酸+高氯酸是常用的消解试剂组合,但是发烟快、易蒸干,高氯酸有爆炸的危险。条件2) 中加入硫酸,可以延长蒸干时间,充分发挥高氯酸的氧化性,提高消解效率,避免发生爆炸;样品中的有机物先被硫酸碳化,再被高氯酸氧化去除。条件2) 试剂用量小,时间短,故选择该法消解样品。
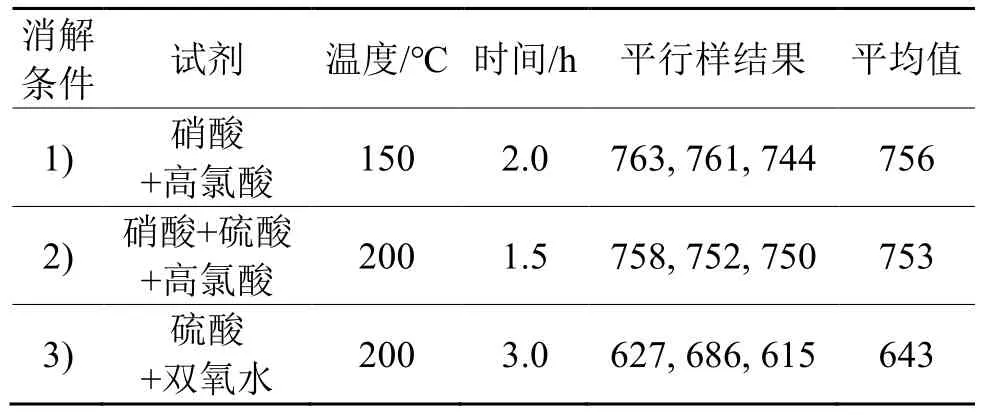
表3 不同消解条件的比较Tab. 3 Comparition of different digestion conditions /(g/t)
2.3 ICP-AES 测定精度的内标法改进
根据铑派克样品铑含量高低不同,以SF03130-1#,sy-1#两个样品为代表,分别称取9 份平行样,按实验方法处理,间隔1 天进行第二次测定,分别用内标法和常规法计算平均值的相对标准偏差(RSD),结果列于表4。

表4 两种方法测定精密度的比较Tab. 4 Comparition of measurement precision by two methods
由表4 可以看出,含铑量越高,RSD越小,即测定精密度越高。对两个不同样品,内标法测定的RSD仅为常规方法的一半左右,具有更好的测定精密度。内标法两次测定结果的偏差明显小于常规方法,具有更好的再现性。
相比常规法,内标法对两个样品两次测定的再现性和精密度均明显改善。原因在于,湿法消解后的铑派克样品试液中共存磷、硫、氯和未完全分解的有机物,不同平行样试液其浓度存在差异,ICP-AES 测定时引起进样速率、雾化效率及激发效率的变化,导致谱线强度波动,精密度变差。加入钇作内标,分析线与内标线的谱线强度同向波动,通过内标线变化对分析线的变化进行补偿和修正,从而改善样品测定的再现性和精密度。
2.4 方法准确度
用加标回收试验验证方法准确度。称取3 份湿法油相SF03130-1#样品(参考值360 g/t),分别加入2.0 mL,5.0 mL,10.0 mL 铑标准溶液,按实验方法处理后测定铑,结果如表5 所示。得到的加标回收率在99.1%~99.7%之间,变化幅度仅为常规方法(97.7%~100.0%)的一半左右,表明内标法具有更高的测定准确度。

表5 加标回收试验Tab. 5 Recoveries of standard addition
2.5 实际样品分析
本方法已用于不同含量铑派克废料实际样品分析,结果见表6,内标法测定结果精密度较常规法测定结果精密度有明显的改善。

表6 铑派克样品测定结果Tab. 6 Determination results of rhdium parker sample
3 结论
1) 采用硝酸+硫酸+高氯酸冒烟消解铑派克样品,2 h 内可完成样品消解,便于后续测定;相比之下,火试金富集称样量大,处理流程长。
2) 在试液和校准曲线溶液中加入定量钇作为内标,用ICP-AES 法测定铑派克废料中的铑,相比不加内标的样品,RSD降低一半左右,加标回收率为99.1%~99.7%,测定精密度、再现性和准确度均明显提升。
3) 对含铑量为100~1500 g/t 的样品,测定相对标准偏差(RSD)均<1.5%,方法准确、快速,已应用于生产分析。
猜你喜欢
汽车工艺与材料(2022年7期)2022-07-21
人民交通(2022年2期)2022-02-28
锻造与冲压(2021年12期)2021-07-01
模具制造(2019年7期)2019-09-25
今日农业(2019年11期)2019-08-13
科学与财富(2018年26期)2018-10-24
中国科技纵横(2018年10期)2018-07-27
智富时代(2018年11期)2018-01-15
智富时代(2018年11期)2018-01-15
课外生活(小学1-3年级)(2017年11期)2017-12-11
