
Lycium barbarum extract promotes M2 polarization and reduces oligomeric amyloid-β-induced inflammatory reactions in microglial cells
2022-06-13 08:33:14ZhongQingSunJinFengLiuWeiLuoChingHinWongKwokFaiSoYongHuKinChiu
中国神经再生研究(英文版) 2022年1期
Zhong-Qing Sun, Jin-Feng Liu, Wei Luo, Ching-Hin Wong, Kwok-Fai So, ,Yong Hu, Kin Chiu,
Abstract Lycium barbarum (LB) is a traditional Chinese medicine that has been demonstrated to exhibit a wide variety of biological functions, such as antioxidation, neuroprotection, and immune modulation. One of the main mechanisms of Alzheimer’s disease is that microglia activated by amyloid beta (Aβ) transform from the resting state to an M1 state and release pro-inflammatory cytokines to the surrounding environment.In the present study, immortalized microglial cells were pretreated with L. barbarum extract for 1 hour and then treated with oligomeric Aβ for 23 hours. The results showed that LB extract significantly increased the survival of oligomeric Aβ-induced microglial cells, downregulated the expression of M1 pro-inflammatory markers (inducible nitric oxide synthase, tumor necrosis factor α, interleukin-6, and interleukin-1β),and upregulated the expression of M2 anti-inflammatory markers (arginase-1, chitinase-like protein 3, and interleukin-4). LB extract also inhibited the oligomeric Aβ-induced secretion of tumor necrosis factor α, interleukin-6, and interleukin-1β in microglial cells. The results of in vitro cytological experiments suggest that, in microglial cells, LB extract can inhibit oligomeric Aβ-induced M1 polarization and concomitant inflammatory reactions, and promote M2 polarization.
Key Words: Alzheimer’s disease; amyloid-β; anti-inflammatory factors; Lycium barbarum extract; M1 microglia; M2 microglia;neuroinflammation; proinflammatory factors
Introduction
Microglial cells play a major role in the innate immune response of the central nervous system. There are two distinct phenotypes of activated microglia: classical microglia(M1, proinflammatory) and alternative microglia (M2, antiinflammatory) (El-Shimy et al., 2015; Lan et al., 2017; Gray et al., 2020). M1 microglia in the classical activation state release pro-inflammatory and neurotoxic mediators that result in neurodegeneration. In contrast, M2 microglia in the alternative activation state deliver anti-inflammatory cytokines and neurotrophic factors that contribute to the process of neural repair and regeneration (Li et al., 2007; Chiu et al.,2009). Whether microglia engender neuroprotection (M2) or neural injury (M1) depends on the nature of the disease itself or the type of stimulation.
Increasing evidence fromin vitroandin vivostudies has indicated that aggregated amyloid beta (Aβ) can activate microglia to release pro-inflammatory cytokines and chemokines, which in turn trigger the inflammatory response in Alzheimer’s disease (AD) (El-Shimy et al., 2015; Long and Holtzman, 2019; Leng and Edison, 2020). The formation of highly neurotoxic oligomeric Aβ (o-Aβ) can cause M1 polarization and the release of neurotoxic molecules, such as superoxide–nitric oxide, tumor necrosis factor alpha (TNF-α),and interleukin 1 beta (IL-1β) (He et al., 2011; Hu et al., 2017).In contrast, M2 polarized microglia exhibit neuroprotective effects in the brain by eliminating Aβ plaques and supporting network remodeling and trophic factors (Sarlus and Heneka,2017; Leng and Edison, 2020). Thus, modulating microglia polarization to induce neuroprotective effects and attenuate neurotoxic inflammatory responses is a novel and promising therapeutic strategy for AD.
Lycium barbarum(LB; also known as goji berry, wolfberry,or gouqizi in Chinese) is a popular, affordable traditional Chinese medicine that has been used for thousands of years(Manthey et al., 2017). In the past decade, studies of the effects of LB in the brain, retina, liver, and systemic immune system have been performed bothin vitroandin vivousing modern research methods (Li et al., 2013; Wang et al., 2014;Zhang et al., 2015; Xing et al., 2016; Manthey et al., 2018;Mi et al., 2020). In AD research, LB polysaccharide (LBP)has been reported to attenuate Aβ-induced neurotoxicity in primary cortical neurons (Ho et al., 2007, 2010), and enhances neurogenesis, ameliorates amyloid pathology, and improves cognitive function in the Aβ precursor protein (APP)/presenilin 1 (PS) transgenic mouse (Zhou et al., 2020). LBP also exhibits an anti-inflammatory effect, reducing the production of inflammatory mediators through the inhibition of nuclear factor kappa B (NF-κB) in a lipopolysaccharide-treated cell line, retinal degeneration 10 (RD10) mouse retinal model, and liver injury model (Xiao et al., 2012; Teng et al., 2013; Wang et al., 2014; Zhang et al., 2015; Manthey et al., 2017). Our previous study in a rat glaucoma model demonstrated that oral feeding of LBP protects the survival of retinal ganglion cells and modulates retinal microglia to an activated status with neuroprotective effects (Chiu et al., 2009). However, it remains to be investigated whether LBP modulates retinal microglia status relating to M1 or M2.
In the present study, we aimed to characterize o-Aβ-induced M1 polarization in an immortalized microglial cell line (IMG)and evaluated whether LB extract (LBE) was able to shift microglia activation from M1 to M2, thereby providing a neuroprotective environment.
Materials and Methods
LBE preparation for cell culture
LBE was prepared and provided by Eu Yan Sang (HK) Ltd. (Hong Kong, China). The dried mature fruit of LB was the product of Zhongning in the Ningxia Hui Autonomous Region, China.The extraction method was as follows: 2.5 kg of LB fruit was washed, soaked in warm water (40°C) for 15 minutes, and boiled for 1 hour. The filtered drug residue was then boiled again. The extract, obtained in two batches, was combined and concentrated to 1.25 kg. Next, the LBE was weighed and diluted in ultrapure water (w/v) to make a stock solution (100 mg/mL).Before use, the LBE was filtered with a 0.22 µm filter membrane to obtain the final solution. Various concentration of LBE were used in the cell culture experiments, and were freshly prepared using Dulbecco’s modified Eagle’s medium (DMEM) low glucose(Thermo Fisher Scientific, Waltham, MA, USA).
Preparation of Aβ1–42 solution
The oligomeric Aβ1–42peptide (the ERI Amyloid Laboratory LLC, Oxford, CT, USA) was prepared according to a previous protocol (Michelucci et al., 2009). Briefly, Aβ1–42peptide was dissolved to 1 mM in 1,1,1,3,3,3-hexafluoro-2-propanol(HFIP; Sigma-Aldrich, St. Louis, MO, USA) and placed in 1.5 mL Eppendorf tubes. The tubes were then left in a biological safety cabinet with the lids open overnight to remove the HFIP.The dried peptide film that remained in the tube was then stored at –80°C for several months. Next, the peptide film was resuspended in anhydrous dimethyl sulfoxide (Sigma-Aldrich)to construct a 5 mM stock solution. For the oligomerization of Aβ, the stock solution was further diluted to 100 µM in DMEM without phenol red. After being incubated for 24 hours at 4°C,the solution was centrifuged at 15,000 ×gfor 10 minutes to harvest the supernatant, which was enriched in Aβ1–42peptide oligomers and was subsequently used in the study. HFIP/dimethyl sulfoxide and dimethyl sulfoxide were used as vehicle controls. The successful oligomerization of Aβ1–42was validated with Tris-Tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot (Additional Figure 1).
Cell culture
IMG cells were purchased from Sigma-Aldrich (Cat# SCC134).This cell line is derived from an adult mouse brain infected with the v-raf/v-myc retrovirus (McCarthy et al., 2016). The IMG cells were cultured in DMEM high glucose medium supplemented with 10% (v/v) fetal bovine serum (Invitrogen,Carlsbad, CA, USA) at 37°C in a humidified atmosphere with 95% air and 5% CO2. When the cells reached approximately 80% confluence, the IMG cells were subcultured onto other coverslips for further studies. IMG cells in passages 4–7 were used in this study.
Assessment of lactate dehydrogenase release
Lactate dehydrogenase (LDH) released into the culture medium was detected using a commercially available assay kit (Cat# 88953; Thermo Fisher Scientific). IMG cells were seeded into 96-well plates with DMEM low glucose medium supplemented with 1% (v/v) fetal bovine serum at a density of 1 × 104cells/well and incubated overnight. The cells were then treated with LBE at concentrations of 100, 250, 500, and 1000µg/mL for 1 hour, followed by treatment with 5 µM oligomeric Aβ1–42for 23 hours. The medium was then transferred to another 96-well plate and mixed with LDH detection buffer.Thirty minutes later, stop solution was added to the wells and the LDH reaction activity was subsequently assessed via spectrometry (EnSpire Multimode Plate Reader; PerkinElmer,Waltham, MA, USA) based on the absorbance at 490 nm wavelength. The ratio of LDH activity to that of control cells was calculated and presented as the percentage of cell death.
PrestoBlue assay
Cell viability was measured with a PrestoBlue assay kit (Cat#A13261; Thermo Fisher Scientific). The IMG cells were seeded in 96-well plates overnight. Next, the cells were treated with LBE at concentrations of 100, 250, 500, and 1000 µg/mL for 1 hour followed by treatment with 5 µM oligomeric Aβ1–42for 23 hours. Cells were then incubated with fetal bovine serum-free medium containing PrestoBlue for 2 hours at 37°C. Finally,index cell viability activity was assessed via spectrometry based on the absorbance at 570 nm.
Immunofluorescence detection
After treatment, the IMG cells were fixed with 4%formaldehyde, blocked with 5% donkey serum (Invitrogen)in phosphate-buffered saline (PBS), and then incubated at 4°C overnight with the following primary antibodies: antirabbit ionized calcium-binding adapter molecule 1 (Iba-1,1:500, Cat# 019-19741; WAKO, Osaka, Japan), anti-rabbit P2Y metabotropic G-protein-coupled purinergic receptors 12 (P2Y12, 1:500, Cat# ab184411; Abcam, Cambridge, UK),anti-rabbit transmembrane protein 119 (TMEM119, 1:500,Cat# ab209064; Abcam), anti-rabbit inducible nitric oxide synthase (iNOS, 1:500, Cat# 13120; Cell Signaling Technology,Beverly, MA, USA), anti-mouse ARG-1 (1:500, Cat# 93668;Cell Signaling Technology), and anti-mouse IL-1β (1:500, Cat#12242; Cell Signaling Technology). The next day, cells were washed three times with PBS and incubated for 2 hours at room temperature with the following secondary antibodies:donkey anti-mouse IgG H&L (Alexa Fluor 568) (1:500, Cat#AB_2534013; Invitrogen), donkey anti-rabbit IgG H&L (Alexa Fluor 568) (1:500, Cat# AB_2534017; Invitrogen), and donkey anti-rabbit IgG H&L (Alexa Fluor 488) (1:500, Cat#AB_2535792; Invitrogen). 4′,6-Diamidino-2-phenylindole(Sigma-Aldrich) was used to stain nuclei. For identified microglia markers, the PBS control was used in place of the primary antibodies. For microglia polarizing markers, the same antibodies were added to all the group and cells without any treatment as blank control. Images were taken using an LSM 710 confocal microscope (Zeiss, Oberkochen, Germany).
Quantitative real-time polymerase chain reaction analysis
Total RNA of IMG cells under different treatment conditions was extracted using an RNA extraction kit (Cat# 74106;QIAGEN, Hilden, Germany) and then reverse transcribed to cDNA using a commercially available kit (Cat# 205413;QIAGEN). To detect the expression of the genes encoding iNOS,TNF-α, IL-1β, ARG-1, IL-4, and Ym-1 (all primer sequences are listed inTable 1), real-time polymerase chain reaction was performed using a SYBR Green Polymerase Chain Reaction Kit(Cat# 208056; QIAGEN). Amplification was performed using an ECO system (Gene Company Limited, Hong Kong, China), with a pre-denaturization step for 30 seconds at 95°C, followed by 40 cycles of 5 seconds at 95°C and 15 seconds at 60°C. The mRNA expression levels of cytokines were normalized to those of β-actin. The results of the treatment groups were expressed as relative changes compared with the average signals obtained from the untreated controls.
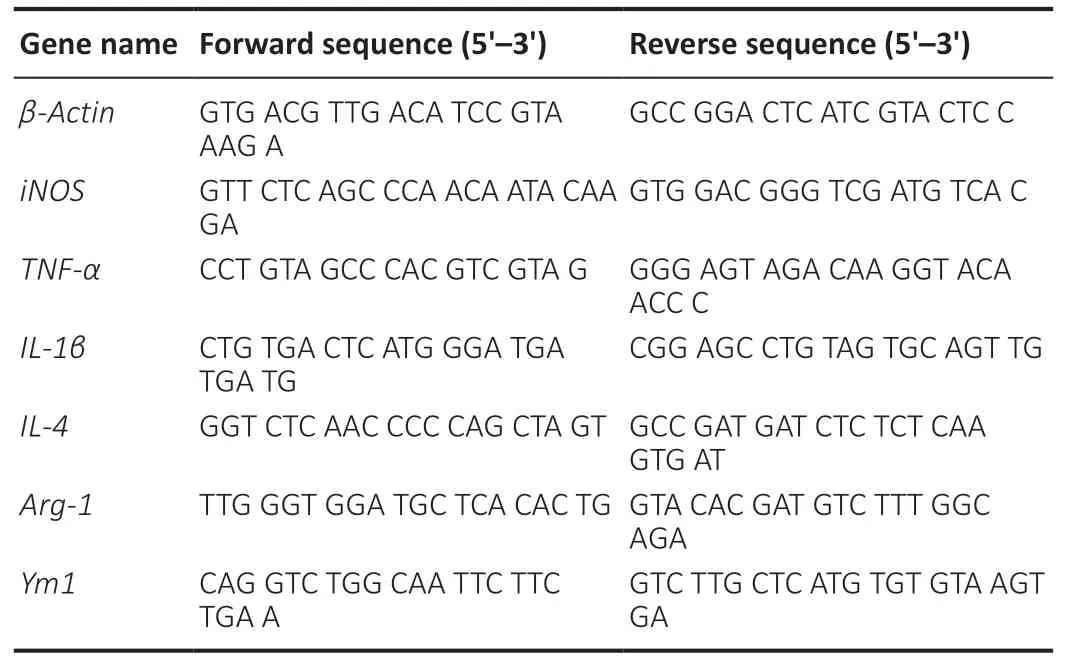
Table 1 |Primer sequences used for quantitative real-time polymerase chain reaction analysis
Western blot analysis
Total protein was extracted from IMG cells using cell lysis buffer, and the concentration was quantified using a bicinchoninic acid assay kit (Cat# 23227; Thermo Fisher Scientific). Next, 20 µg protein was loaded and segregated on a 10% sodium dodecyl sulfate-polyacrylamide gel before being transferred onto polyvinylidene fluoride membranes(Millipore, Burlington, MA, USA) for blotting. The membrane was blocked at room temperature for 1 hour with fresh blocking buffer containing 5% skim milk powder, and was then incubated at 4°C overnight with the following primary antibodies: iNOS (anti-rabbit, 1:1000, Cat# 13120; Cell Signaling Technology), TNF-α (anti-rabbit, 1:1000, Cat# 6954;Cell Signaling Technology), IL-1β (anti-mouse, 1:500, Cat#12242; Cell Signaling Technology), ARG-1 (anti-rabbit, 1:500,Cat# 93668; Cell Signaling Technology), IL-6 (anti-rabbit, 1:500,Cat# 12912; Cell Signaling Technology), and β-actin (antimouse, 1:1000, Cat# 3700; Cell Signaling Technology). The membrane was then washed with Tris-buffered saline with Tween-20 and incubated for 2 hours at room temperature with the following secondary antibodies: goat anti-mouse IgG(H + L) horseradish peroxidase (1:5000, Cat# AB_2534060;Invitrogen) and goat anti-rabbit IgG H&L (Alexa Fluor 568)(1:5000, Cat# AB_143157; Invitrogen). The blots were visualized using an enhanced chemiluminescence system(Millipore) and the images were captured using a gel imager(Bio-Rad, Hercules, CA, USA). The optical density value of each band was measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The relative expression of each immunoreactive band was calculated by comparing the target protein band with that of β-actin.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay kits (R & D Systems,Minneapolis, MN, USA) were used to evaluate the expression of TNF-α, IL-1β, and IL-6 upon o-Aβ1–42exposure. After treatment, the levels of these proteins in the supernatant of cultured IMG cells were measured using enzyme-linked immunosorbent assay. The chromogenic reaction was monitored by detecting the absorbance at 450 nm with an EnSpire Multimode Plate Reader.
Statistical analysis
Results were statistically a nalyzed using one-way analysis of variance followed by Tukey’spost hoctests and the Student’st-test in Prism GraphPad 5 (GraphPad, La Jolla, CA, USA).Data are presented as the mean ± standard deviation (SD).In all analyses, aP-value of less than 0.05 was considered statistically significant.
Results
Microglial markers are detected in IMG cells
IMG cells were identified by the widely used microglia markers Iba-1, P2Y12, and TMEM119. Iba-1, P2Y12, and TMEM119 were all detected in the cytoplasm of cultured cells, while no staining was detected in the PBS negative control sections(Figure 1). IMG cells were therefore qualified for use in the subsequent experiments.
LBE prevents o-Aβ-induced cytotoxicity in IMG cells
To determine effective concentrations, IMG cells were treated with different concentrations of o-Aβ or LBE for 24 hours, and the LDH released into the conditioned media by the treated cells was measured as an indicator of cellular cytotoxicity. In addition, the adherent IMG cells were further incubated with PrestoBlue reagent to examine cell viability. Compared with the control group, a dose-dependent increment in cytotoxicity caused by o-Aβ was observed when the concentration surpassed 2.5 µM (P< 0.01;Figure 2A). This was accompanied by a marked decrease in cell survival starting from 5 µM o-Aβ(P< 0.01;Figure 2B). These results indicated that 5 µM o-Aβ was the optimal dose for studying the effects of LBE on IMG cells. In addition, IMG cells were stimulated with various concentrations of LBE, and a significant level of cytotoxicity was observed from 500 µg/mL (P< 0.001;Figure 2C). Moreover,a decline in cell viability was observed at 1000 µg/mL LBE (P< 0.01;Figure 2D).
Next, a co-treatment strategy was adopted to investigate the effects of LBE on o-Aβ-induced cellular cytotoxicity and viability. Pretreatment with LBE for 1 hour followed by treatment with 5 µM o-Aβ and various concentrations of LBE for an additional 23 hours indicated that 100–250 µg/mL of LBE was sufficient to prevent o-Aβ induced cytotoxicity in IMG cells (P< 0.05;Figure 3A). Furthermore, a comparable increase in cell survival was observed with LBE pretreatment compared with o-Aβ treatment alone (P< 0.05;Figure 3B).Together, these results suggest that LBE can inhibit the neurotoxicity induced by o-Aβ and exerts a certain protective effect in IMG cells.
LBE alters o-Aβ-induced microglia polarizing markers
To better understand the role of LBE in the context of diseaseassociated microglia, immunostaining was performed for pro- and anti-inflammatory markers in IMG cells (Figure 4).O-Aβ treatment alone increased the immunoreactivity of the M1 pro-inflammatory markers iNOS and IL-1β (Figure 4A;middle panel) compared with the control group. In contrast,LBE pretreatment (250 µg/mL) significantly reduced the immunoreactivity of iNOS and IL-1β (P< 0.01;Figure 4A–C)and significantly upregulated the M2 anti-inflammatory marker ARG-1 (P< 0.01;Figure 4DandE). This immunoreactivity profile suggests a polarizing effect of LBE pretreatment that drives the microglial activation state toward the antiinflammatory (neuroprotective) M2 type and away from the pro-inflammatory (neurotoxic) M1 type. These results indicate the potential role of LBE in suppressing inflammatory cascades and facilitating microglial neuroprotection.
LBE suppresses the pro-inflammatory response and promotes microglial anti-inflammatory properties
To further explore the IMG cell polarizing patterns that were observed using immunostaining, total RNA was extracted for semi-quantitative real-time polymerase chain reaction.Compared with the control group, 5 µM o-Aβ treatment led to significantly elevated mRNA levels of the pro-inflammatory markersTNF-α,IL-1β,IL-6, andiNOS(P< 0.05;Figure 5A–C). In contrast, LBE pretreatment significantly reduced these elevated marker levels. We then measured the effects of LBE pretreatment by quantifying the mRNA expression of antiinflammatory markers. Treatment with o-Aβ downregulated the mRNA expression ofARG-1andYm1compared with the control group. LBE pretreatment upregulated the mRNA expression ofIL-4,ARG-1, andYm1(P< 0.05;Figure 5D–F)compared with o-Aβ treatment alone. Together, these findings suggest that LBE inhibits the o-Aβ-induced pro-inflammatory response and promotes anti-inflammatory properties.
LBE inhibits inflammatory protein synthesis in o-Aβ-treated IMG cells
Treatment with o-Aβ increased the protein levels of several pro-inflammatory factors, including iNOS, TNF-α, IL-1β, and IL-6, all of which were significantly inhibited by LBE pretreatment(P< 0.05;Figure 6A–D). LBE pretreatment also elevated the expression of the anti-inflammatory factor ARG-1 (P< 0.05;Figure 6E).
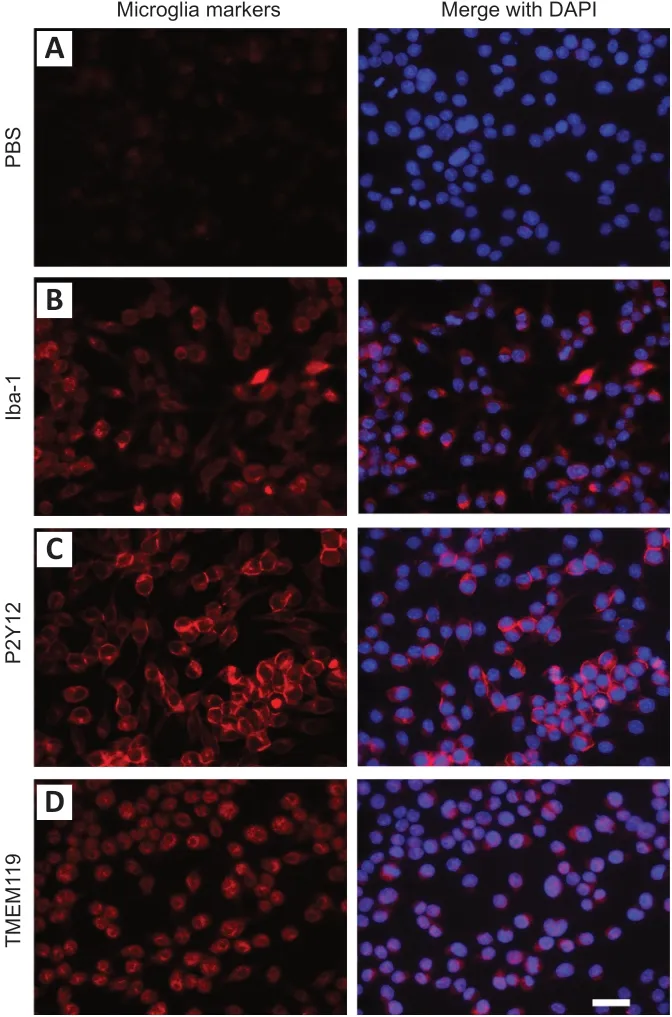
Figure 1|IMG cells were identified by different microglia markers.
LBE affects o-Aβ-induced inflammatory cytokine secretion in IMG cells
The successful modulatory effect of LBE likely stemmed from its compensatory effects on mRNA- and protein-level alterations by modulating the release of pro-inflammatory factors. To confirm this notion, profiles of the cytokines released into culture supernatants from IMG cells were investigated using enzyme-linked immunosorbent assay(Figure 7). Compared with o-Aβ treatment, LBE pretreatment significantly reduced the secretion of pro-inflammatory factors, such as TNF-α, IL-1β, and IL-6 (P< 0.05;Figure 7A–C).These results further support the finding that LBE treatment can inhibit o-Aβ-induced production of pro-inflammatory factors and neurotoxic secretion in IMG cells.
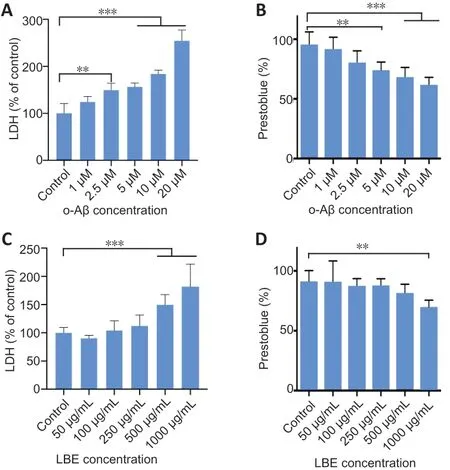
Figure 2|LDH and PrestoBlue assay responses to o-Aβ or LBE treatment in IMG cells.
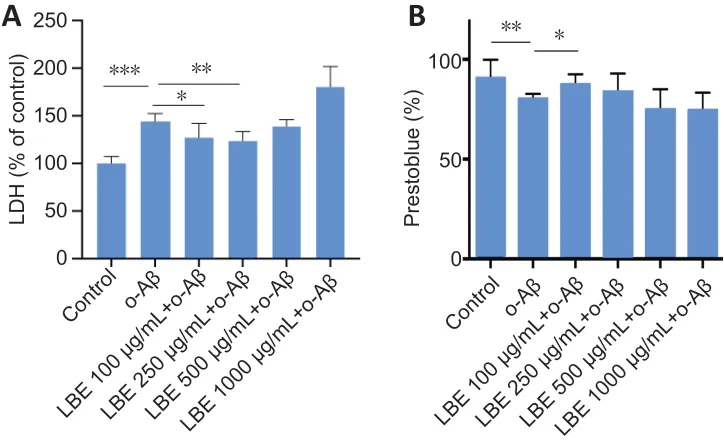
Figure 3|Effects of o-Aβ and LBE on IMG cell cytotoxicity and viability.
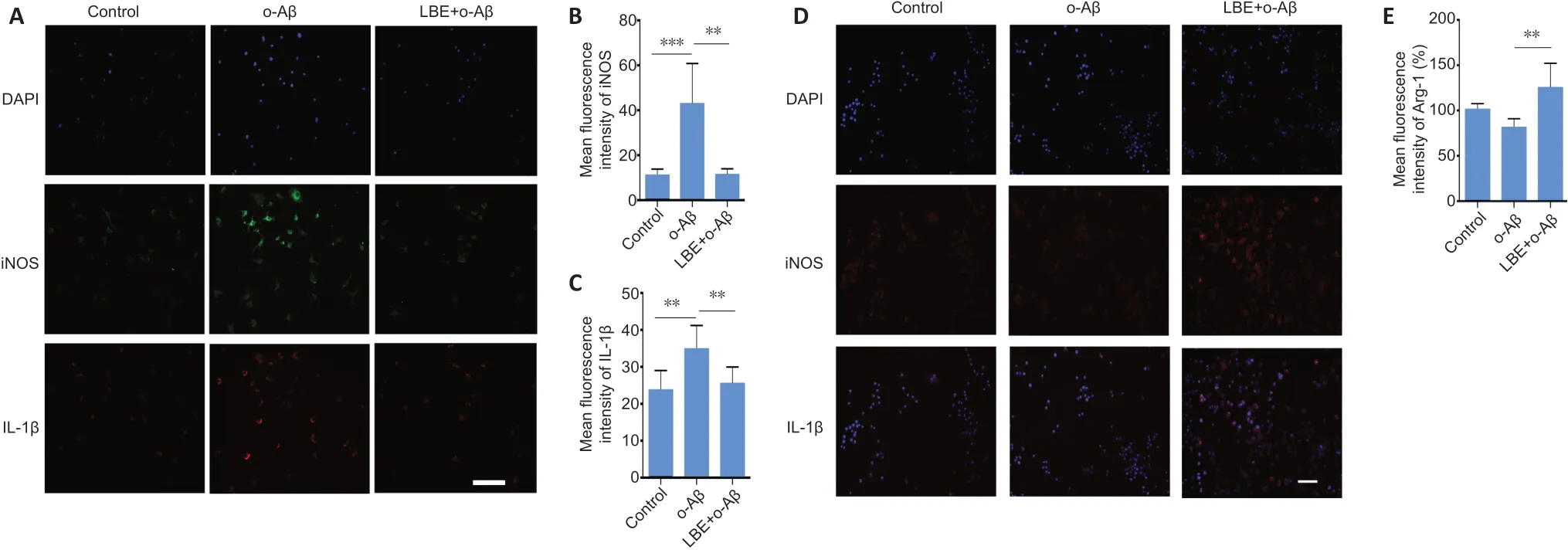
Figure 4|LBE pretreatment alters o-Aβ-induced microglia polarizing markers.
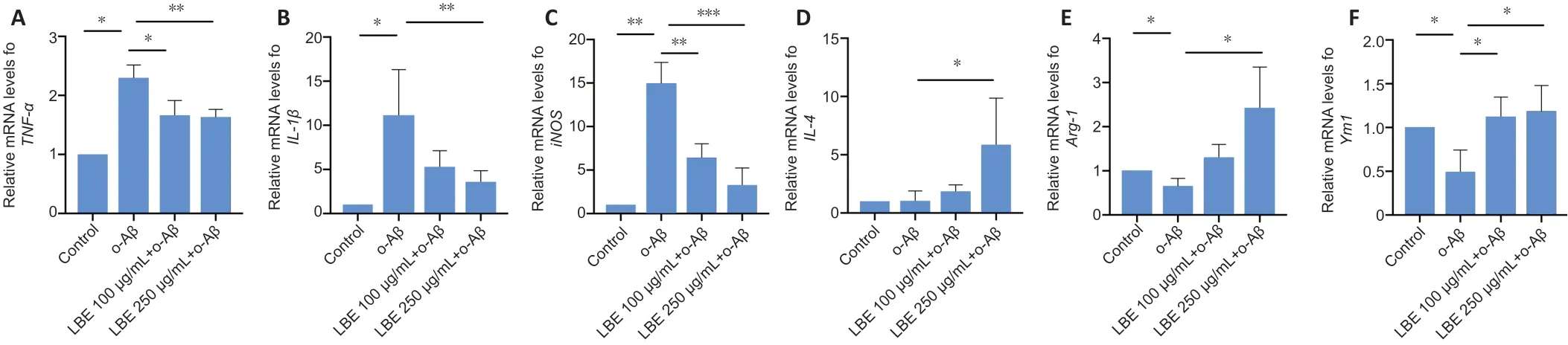
Figure 5|Effects of LBE on the o-Aβ-induced mRNA expression of inflammatory cytokines in IMG cells.
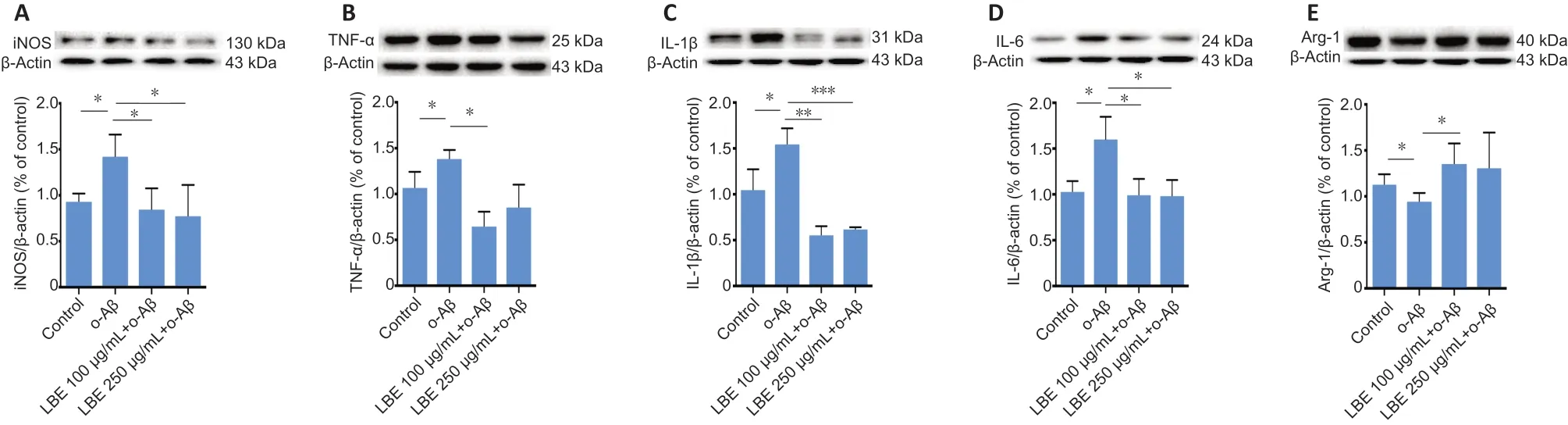
Figure 6|LBE suppresses o-Aβ-induced inflammatory protein synthesis in IMG cells.
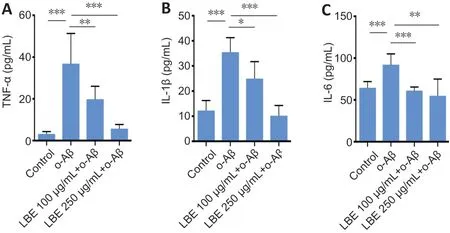
Figure 7|Effects of LBE on o-Aβ-induced inflammatory cytokine secretion in IMG cells.
Discussion
Neuroinflammatio n mediated by microglia is a key factor in the pathogenesis of AD (Sarlus and Heneka, 2017). In our study, o-Aβ elicited an M1 microglial response without significantly compromising cell viability. LBE may inhibit the M1 phenotype induced by o-Aβ and promote an M2 state in IMG cells.
The age-dependent Aβ pathological process is associated with the microglial activation phenotype in the hippocampus of PS1M146L/APP751SLdouble-transgenic AD mice (Jimenez et al., 2008). In these mice, the M2 microglial phenotype (Ym1-positive and TNF-α-negative) is restricted to Aβ plaques at 6 months of age, while microglial activation spreads to the entire hippocampus area by 18 months of age, indicating a classic M1 phenotype (with expression of TNF-α, COX2,and NOX1), with accumulated levels of soluble Aβ oligomers(Jimenez et al., 2008; Tang and Le, 2016). These results indicate that M2 microglia might intend to phagocytose the Aβ plaques at 6 months. M2 microglia possess neuroprotective functions, remove Aβ plaques, and enhance phagocytosis,resulting in improved cognitive function (Li et al., 2007; Tang and Le, 2016; Sarlus and Heneka, 2017). When Aβ plaques accumulate to an amount that is beyond the capacity of microglial phagocytosis, M2 microglia may shift to an M1 phenotype and become detrimental to the surrounding neurons. As shown in primary microglia cells, o-Aβ provokes microglia from the resting state to the M1 activation state and enhances the release of cytotoxic and inflammatory factors(Michelucci et al., 2009). This might explain the observation of M1 microglia in the hippocampus of PS1M146L/APP751SLtransgenic AD mice at 18 months. Thus, any protocol that can shift microglia from an M1 to an M2 state might provide neuroprotection and improve cognitive function in AD mice.Such an M1 to M2 shift has been demonstrated in Aβ-stressed primary microglial cells. IL-4, an anti-inflammatory cytokine,can potently downregulate the expression of IL-6 and TNF-α and upregulate the transcription levels of C-C chemokine receptor type 2 and ARG-1 in cells under Aβ stress (Lyons et al., 2007).
Similarly, Chinese medicine extracts have also been shown to suppress M1 microglial activity and reduce inflammatory cytokine levels. For example, resveratrol has anti-inflammatory effects via the suppression of M1 microglia activation and the amelioration of proinflammatory cytokine release (Gomes et al., 2018). Furthermore, not only can resveratrol significantly inhibit the Aβ1–42-induced expression of the proinflammatory cytokines IL-6, IL-1β, and TNF-α in BV2 cells (Feng and Zhang,2019), but it can also restore spatial memory and protect adult Sprague Dawley rats from Aβ-induced neurotoxicity by suppressing iNOS production (Zhao et al., 2018). In addition,curcumin, the major yellow pigment in turmeric, improves primary microglia viability against fibril Aβ1–42and significantly reduces the expression of pro-inflammatory factors, including TNF-α, IL-1β, and IL-6 (Shi et al., 2015). Moreover, Teter et al. (2019) revealed that low-dose curcumin downregulates CD33 and upregulates TREM2 expression (thus decreasing AD risk), and suppresses the M1 microglia cells that decrease the expression of proinflammatory factors (CD11b, iNOS, COX2,and IL-1β) in APPswe transgenic mice.
In the current study, LBE simultaneously promoted the expression of anti-inflammatory ARG-1 and inhibited o-Aβinduced overexpression of proinflammatory iNOS and IL-1β.Western blot and real-time polymerase chain reaction results further confirmed that LBE can both reduce the expression of M1 markers (iNOS, TNF-α, IL-6, and IL-1β) and increase the expression of M2 markers (ARG-1 and Ym1). These results indicate that LBE can attenuate the o-Aβ-induced neuroinflammatory response of microglia by reducing the release of inflammatory factors and promoting the release of anti-inflammatory factors, thereby reversing the activation state of microglia from M1 to M2. Although there were some limitations to our study—such as it being a cell line study, with no interference of inhibitors, and using mixture compounds of LBE—we propose that our study was meaningful because we provided a new potential target for preventing AD. Future studies in AD animal models should be carried out to explore the M1 and M2 states after LBE treatment.
LBE has been reported to stabilize the blood barriers in the central nervous system in various preclinical studies.In a mouse model of cerebral ischemic stroke, LBE was demonstrated to effectively improve neurological function recovery, reduce the infarct area, and protect the brain from blood–brain barrier disruption (Yang et al., 2012). Additionally,pretreatment with LBP for 1 week reportedly protects the survival of retinal ganglion cells in retinal ischemia/reperfusion injury, reduces oxidative stress, inhibits the activation of glial cells, and blocks the destruction of the blood–retinal barrier(Li et al., 2011). Thus, stabilization of the blood–brain barrier,direct neuroprotection, and switching of microglia from M1 to M2 are the multi-target functions of LBE. These functions and the affordable price of LB may make it valuable for use in AD prevention and treatment. In summary, pretreating IMG microglial cells with LBE promoted M2 polarization and concurrently inhibited Aβ-induced M1 polarization and its associated inflammatory reaction.
Author contributions:Study design: ZQS, KC; experiment implementation:ZQS; data analysis and manuscript drafting: ZQS, WL, JFL; manuscript revision: ZQS, CHW, KC, YH, KFS. All authors have read and approved the final manuscript.
Conflicts of interest:The authors declare that they have no conflict of interest.
Financial support:This work was supported by Midstream Research Program for Universities, Hong Kong Special Administrative Region, China,No. MRP-092-17X. The funding source had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:Not applicable. The IMG cell line used in the current study was purchased from Sigma-Aldrich (SCC134).There is no live animal involved.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Randall L. Davis, Oklahoma State University, USA.
Additional files:
Additional file 1: Open peer review report 1.
Additional Figure 1: Oligomerization of Aβ1–42 was identified by western bolt.

- 中国神经再生研究(英文版)的其它文章
- Inhibition of CXCR4/CXCL12 signaling: a translational perspective for Alzheimer’s disease treatment
- The new wave of p75 neurotrophin receptor targeted therapies
- The anatomical, electrophysiological and histological observations of muscle contraction units in rabbits:a new perspective on nerve injury and regeneration
- Inhibition of LncRNA Vof-16 expression promotes nerve regeneration and functional recovery after spinal cord injury
- Intranasal insulin ameliorates neurological impairment after intracerebral hemorrhage in mice
- Exosomes derived from bone marrow mesenchymal stem cells protect the injured spinal cord by inhibiting pericyte pyroptosis