
面向蛋白质功能预测中有向无环图标记结构的多示例多标记学习
2022-06-08 09:10吴建盛唐诗迪梅德进朱燕翔刁业敏
国防科技大学学报 2022年3期
吴建盛,唐诗迪,梅德进,朱燕翔,刁业敏
(1. 南京邮电大学 地理与生物信息学院, 江苏 南京 210023; 2. 南京邮电大学 通信与信息工程学院, 江苏 南京 210003;3. 南京仁面集成电路技术有限公司, 江苏 南京 210088; 4. 南京叁角加文化发展中心, 江苏 南京 210005)
在经典监督学习中,一个对象仅由一个示例来表示,且仅有一个对应标记。实际上,一个对象可由多个示例来表示,并属于多个类别标记[1],其对应的学习框架叫作多示例多标记学习(multi-instance multi-label learning, MIML)。
过去十几年来,研究者们提出了许多基于MIML的算法,例如MIMLBoost[2]将MIML问题分解为多个单标记多示例子问题单独求解,而MIMLSVM[3]将其分解为多个单示例多标记问题;Yang等提出了一种基于全连接卷积网络的多示例多标记学习方法MIML-FCN+[4];Nguyen等提出了基于深度神经网络的DeepMIML模型[5],为MIML生成表示示例的同时,自动识别与标记相关联的关键示例;Zhu等后来又提出了DMNL[6],用一种高效的增强型拉格朗日优化方法预测隐藏的新颖标记;Hu等提出了一种利用标记相关性的度量多示例多标记学习算法MI(ML)2kNN[7]。
近年来,多示例多标记学习被应用于众多场景中,例如Song等提出了MMCNN-MIML[8],将卷积神经网络(convolutional neural network,CNN)模型引入MIML的图片分类问题中;Xu等将MIML应用于预测肝癌细胞的基因突变问题[9];Li等提出基于卷积神经网络的层次性多示例多标记学习方法HMIML来对果蝇胚胎发育图像进行分类[10];Li等提出的AC-MIMLLN[11]模型将MIML应用于情感分析任务;Mercan等基于MIML方法来对乳腺组织病理图像进行多分类研究[12];Zhang等利用MIML学习并基于时空预修剪技术来对原始视频中的动作进行识别与定位[13];Li等开发了多示例多标记学习网络并应用于情感类别预测[14];Pan等将MIML应用于雷达信号的识别中[15]。
在MIML学习问题里,标记之间往往是相互关联的,其中有向无环图(directed acyclic graph, DAG)是一种常见的层次关联结构,它从任意一个顶点出发均不能经过若干条边回到该顶点。蛋白质常包含多个结构域,也同时拥有多种生物学功能。蛋白质中每个结构域可以独立或与周边结构域相互协作完成其生物学功能。蛋白质生物学功能预测也可表示为多示例多标记学习问题,其中每个蛋白质表示为多MIML学习中的一个样本对象,而每个结构域表示为一个示例,每个生物学功能表示为一个标记[16]。蛋白质的生物学功能有多种描述方式,其中基因本体学(gene ontology,GO)使用最为广泛[17],其中的基因功能本体就是一个DAG结构的典型例子。目前也有不少研究利用GO结构辅助进行生物学功能学习。Li等使用层次聚类的方法,针对GO的结构,在经典的层次聚类模型中加入了新的聚类条件对GO生物学功能进行了预测[18];Zhang等使用了深度神经网络,结合蛋白质序列以及蛋白质-蛋白质结合(protein-protein interaction, PPI)网络,对蛋白质的GO生物学功能进行预测[19];Zhao等引入基于转换器的双向编码表征(bidirectional encoder representation from transformers,BERT)模型从蛋白质的GO标记及其序列中提取特征,对配体-受体结合亲和力(drug-target binding affinity, DTA)进行预测[20]。
目前还没有有效的算法可针对基于DAG结构的MIML问题进行学习。因此,本文提出新的基于有向无环图结构的多示例多标记学习(MIML based on directed acyclic graph, MIMLDAG)算法,通过训练标记共享低维子空间,降低模型排序损失,并融入标记间DAG间层次结构关系对样本预测标记进行优化,提升了算法的学习性能。
1 MIMLDAG算法
提出面向标记之间有向无环图结构的多示例多标记学习算法。算法分三层构建模型,其中前两层充分借鉴了MIMLfast算法[21]的思想。首先,算法从原始数据集的特征空间训练出一个被所有标记共享的低维子空间;然后,训练标记线性模型并通过随机梯度下降方法来优化排序损失;最后,在层次性结构中找到一个子图[22],通过合并子节点来构建与有向无环图结构层次一致的多标记信息,融入标记间的有向无环图结构,得到未见示例样本的层次性标记集合。MIMLDAG算法的整体框架如图1所示,算法伪代码见算法1。
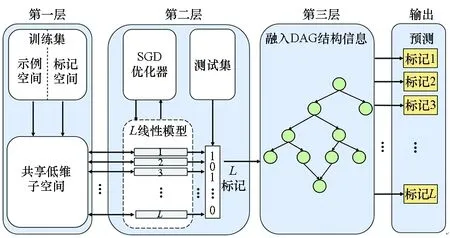
图1 MIMLDAG算法框架Fig.1 Framework of the MIMLDAG algorithm

算法1 MIMLDAG算法伪代码
1.1 训练线性模型
1.1.1 共享低维子空间
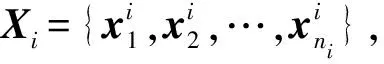
1.1.2 优化排序损失
在第二层中,利用上一层的共享矩阵W0可以将任意样本包Xi的示例x在第l个标记上的分类器定义为:
(1)
式中,wl是第l个标记的m维权重向量。
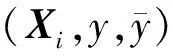
(2)
式中,
(3)

(4)
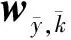
(5)
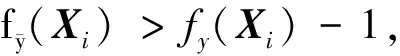
(6)
1.2 融入有向无环图结构
有向无环图结构层次性约束有两种情况[24]:情况A是如果某一节点的标记为正,那么它的所有父节点的标记也为正;情况B是如果某一节点的标记为正,那么它所有父节点中至少有一个节点的标记为正。其中情况A较为常用。对于给定测试样本包XT的预测结果YT={y1,y2,…,yT}∈{0,1},将每个预测标记看作一个节点,已知样本包对应L个标记,那么情况A优化问题表示为:
(7)
其中,若集合YT呈现情况A的层次结构,则称Ψ={Ψ1,Ψ2,…,ΨT}为情况A的nonincreasing集合。
1.3 算法分析
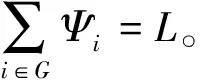
2 算法仿真
2.1 实验数据
使用了三组蛋白质生物学功能预测数据集来对MIMLDAG算法进行实验仿真分析,其分别为G蛋白偶联受体数据集、硫土杆菌(Geobacter sulfurreducens,GS)的蛋白质数据集和古细菌死海盐盒菌(Haloarcula marismortui, HM)的蛋白质数据集 (见表1)。
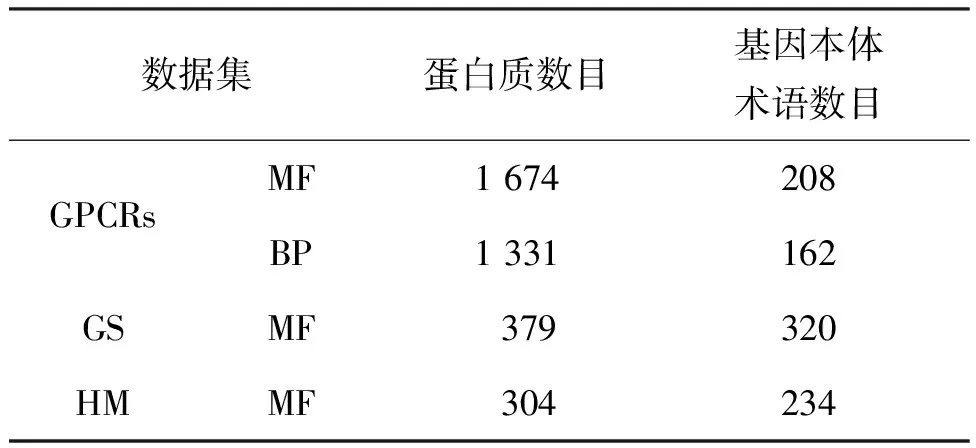
表1 数据集的描述
2.1.1 G蛋白偶联受体数据集
G蛋白偶联受体数据集是从UniProt数据库[25]中下载得到共3 052个G蛋白偶联受体(G protein-coupled receptor,GPCR),再通过UniProt ID号从UniProt数据库中得到所有GPCR的FASTA格式序列。接着,将其输入NCBI的blastclust可执行程序,对GPCR序列进行去冗余处理,将得到的非冗余GPCR样本数据集提交到NCBI的Batch CD-Search服务器,得到GPCR的保守结构域。对于每一个结构域,从以下7个方面构建其特征:
1)三联氨基酸组成(conjoint triad)信息:把20种氨基酸依据其侧链体积与偶极矩分为6类。针对每个结构域,根据其氨基酸序列计算三联体出现频率,其特征维数为216[26]。
2)氨基酸关联(amino acid correlation, AAC)信息:依据上面的6类氨基酸信息来计算每个结构域中两两氨基酸间的AAC信息,对每个结构域,其ACC特征的维数为144[27]。
3)二级结构关联(secondary structure element correlation, SSC)信息:通过PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/ psiform.html)所提供的在线分析工具完成蛋白质二级结构的预测,然后计算3类二级结构类型在结构域中的关联信息SSC(κ),κ∈{2,4,8,16}。对每个结构域,其二级结构关联信息特征的维数为72。
4)进化信息:通过psiblast程序[28]产生位置特异性得分矩阵以表示结构域的进化信息。对于氨基酸长度为n的结构域,其位置特异性得分矩阵的维数为42n。考虑因氨基酸长度不同导致矩阵大小不同的问题,把每个蛋白质的结构域当作一个示例,然后由算法miFV[29]统一为单一向量,对应到单个GPCR结构域的位置特异性得分矩阵特征维数为84。
5)信号肽特征信息(SignalP):通过一种基于神经网络的SignalP 4.0方法[30]从GPCR序列中提取信号肽特征信息。在SignalP 4.0中使用了两种类型的网络,首先使用跨膜数据序列作为负数据来训练得到SignalP-TM网络;然后在缺失这些负数据的情况下训练得到SignalP-noTM网络;最后使用简单的决策方案来选择使用哪个网络:如果SignalP-TM网络预测4个或者更多位置为跨膜位置,则SignalP-TM被用于最终的预测,否则使用SignalP-noTM网络预测。对于每个结构域,SignalP特征维数为84。
6)无序区域特征信息(Disorder):通过DISOPRED 2.43程序[31]预测得到蛋白质的无序区域特征信息。DISOPRED服务器允许用户提交一个蛋白质序列,然后返回每个无序区的无序概率估计值作为无序区域特征信息。对于每个结构域,Disorder特征维数为84。
7)SDK(scientific database maker)软件预测出的特征信息:通过SDK[17]预测蛋白质特征。它从Swiss-Port[32]数据库中提取蛋白质数据,同时对蛋白质进行数据分析,包括物理化学分布计算、同源序列搜索、多序列比对等,得到105维的数据特征。将非数据部分特征去掉,归一化后得到59维的蛋白质数据特征。
最后,对于样本空间中的每个结构域,共有特征维数743。
依据生物学过程与分子功能两个方面来描述蛋白质生物学功能。首先,根据GPCR蛋白质数据集的UniProt ID号从UniProt-GOA ftp站点(ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/)下载得到其对应基因本体学术语(GO terms)ID号及其对应的GO术语;然后,从基因本体学网站(http://geneontology.org/page/download-ontology)下载go.obo文件,分别得到BP与MF的GO术语对应的父节点;最后,基于样本中含有的GO terms及其所有父节点GO terms,构建标记的DAG层次性结构。对于BP,得到非冗余GPCRs样本1 331个,GO术语162个,GO术语的层次性结构深度为9;对于MF,得到非冗余GPCRs样本1 674个,GO术语208个,GO术语的层次性结构深度为12 (见表1)。
2.1.2 GS和HM蛋白质数据集
GS的蛋白质数据集和HM的蛋白质数据集来自http://www.lamda.nju.edu.cn/data_MIMLprotein.ashx[16],此处不再赘述。
2.2 性能比较
与多种经典的多示例多标记学习算法进行比较。这些算法分别为:基于径向基核函数神经网络的MIMLRBF[33]、基于集成学习的EnMIMLNN[16]、基于K邻近算法的MIMLKNN[34]、基于支持向量机的MIMLSVM[3]和快速多示例多标记学习MIMLfast[21]。
对于上述对比算法,本文选用了对应参考文献中的默认参数。其中, MIMLRBF算法的缩放因子为0.08,分数参数为0.1;EnMIMLNN算法中学习率设为0.4;MIMLKNN算法中聚类簇占据样本包的比例为40%;MIMLSVM算法中,高斯核半径r设置为0.2;对MIMLDAG算法,低维共享空间的维度m设置为50;对MIMLfast算法,共享空间维度设为100。
此外,本文采用十倍交叉验证对MIML模型三种常用的评价指标HL(hamming loss)[33]、MaF1(Macro-F1)[35]、MiF1(Micro-F1)[35]进行评估。HL表示样本的预测标记与真实标记之间的错误率。 MaF1先计算在每个标记上的F1值,然后求在所有标记上的平均值,MaF1容易受到样本量少的标记的预测结果影响;MaF1越大,表示模型性能越好。MiF1计算在所有示例包和类别标记上预测结果的F1值;MiF1越大,表示模型性能越好。
2.2.1 性能分析
表2展示了不同数据集下几种算法在不同指标上的性能变化,其中↑表明指标越大模型性能越好,↓反之。由表2可知,对于GPCRs的分子功能MF,相比于其他五种方法,MIMLDAG在HL、MaF1、MiF1上都获得了最好的性能。同样,对于GPCRs的生物学过程,MIMLDAG在HL、MaF1、MiF1方面均取得了最好的性能。对于GS的分子功能,MIMLDAG在MaF1获得了最优的性能。对于HM的分子功能,MIMLDAG在HL上性能较弱,不如EnMIMLNN,也略低于MIMLRBF和MIMLSVM,在其他上都取得了最好的性能。由此可见,MIMLDAG方法比其他多示例多标记学习方法取得了更好的性能。
2.2.2 时间效率分析
表3给出了所有算法在4种数据集上的时间开销。在算法训练及测试时间开销上,对于所有的数据集,MIMLDAG方法的速度与最快的MIMLfast方法基本上接近,明显优于其他4种多示例多标记学习方法。
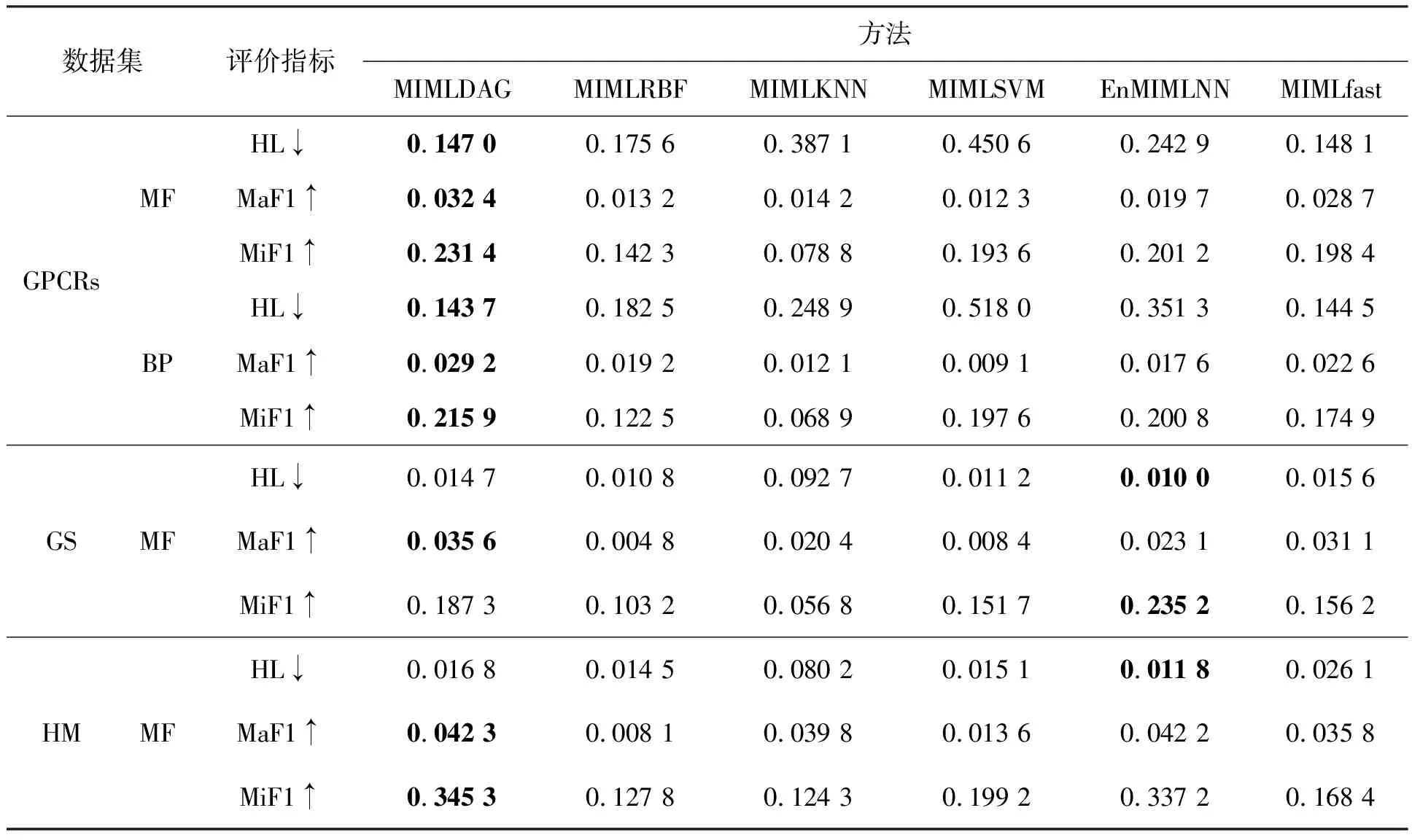
表2 不同数据集上与多示例多标记学习方法的性能比较
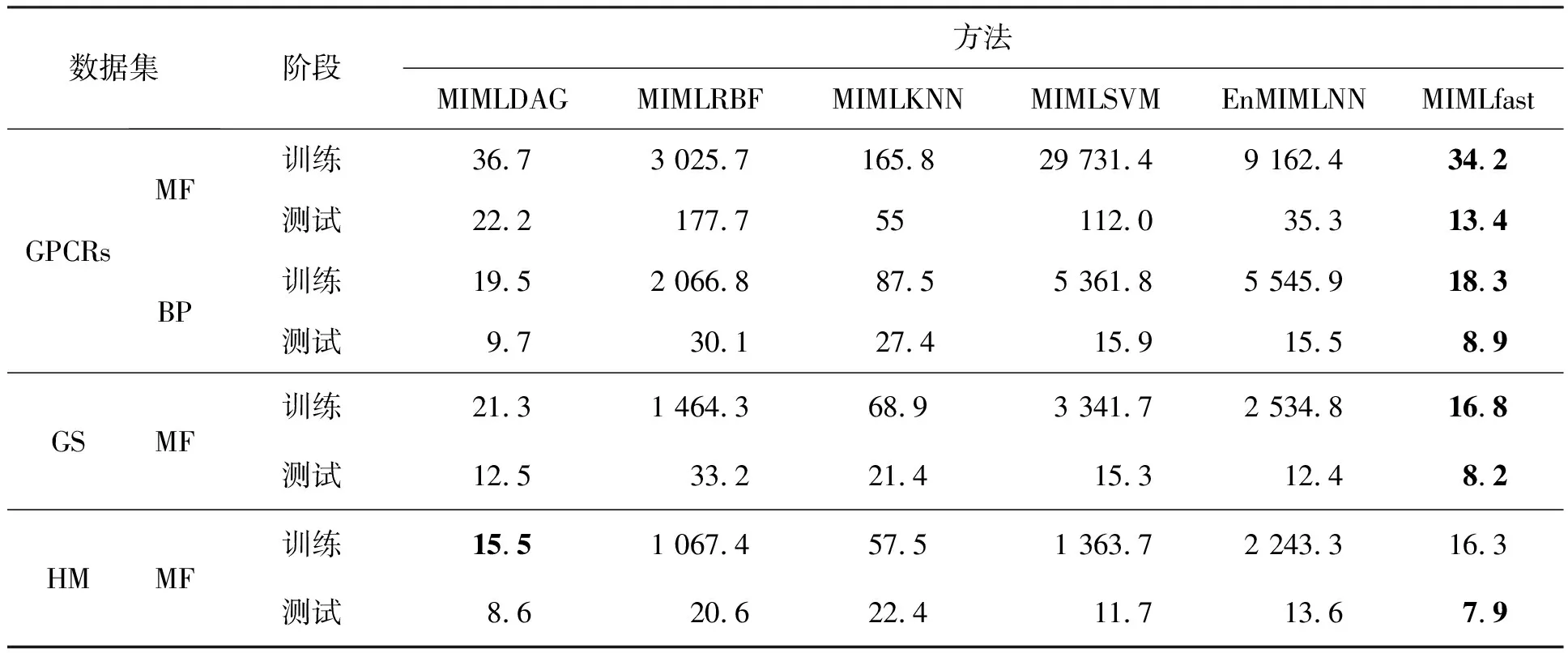
表3 不同数据集上与各多示例多标记学习方法的时间开销比较
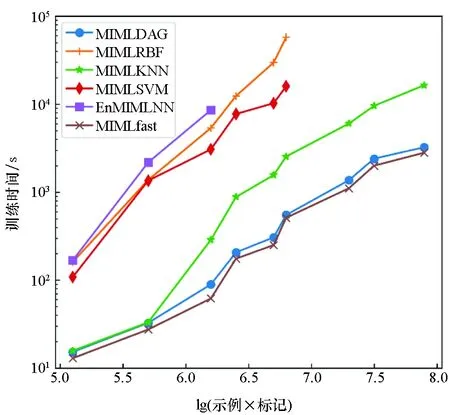
(a) 训练时间(a) Training time
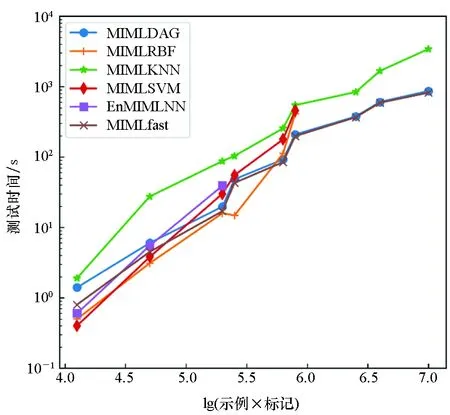
(b) 测试时间(b) Testing time图2 6种MIML算法在不同数量示例和标记上的训练和测试时间开销Fig.2 Runtime of training and testing on six MIML methods with various sizes of instances and labels
图2展示了各个算法在不同数据规模下的时间增长模式。由图可知,对于训练时间,EnMIMLNN的增长速度最快,而MIMLfast和MIMLDAG增长速度较为缓慢;对于测试时间,MIMLKNN算法的增长速度最快,当lg(示例×标记)>6.0时,MIMLDAG的增长速度最为缓慢。因此,MIMLDAG算法随数据集的增长速率很低,基本与MIMLfast方法持平。MIMLDAG算法共分三层:第一层,从原始数据集的特征空间训练出一个被所有标记共享的低维子空间;第二层,训练标记线性模型并通过随机梯度下降方法来优化排序损失;第三层,从层次性结构中找到一个子图,通过合并子节点来构建与有向无环图结构层次一致的多标记信息,得到未见示例样本的层次性标记集合。由式(1)~(6)可以推导出MIMLDAG算法前两层的时间复杂度为Ο[t×L×(d×L)×m],其中t为迭代轮数,L为标记空间中的标记个数,d为示例的特征维度,m为共享低维子空间的维度(m≪d)。算法第三层(算法1步骤7~17)的时间复杂度为Ο[L×log(L)],其中L为标记空间中的标记个数,也就是DAG中的节点数。因为前两层与第三层之间是串行连接,所以算法总的时间复杂度为Ο[t×L×(d×L)×m+L×log(L)]。 可以看出,MIMLDAG算法与样本的数量无关,同时算法将样本原始特征空间映射到低维共享空间,减少了内存消耗并降低了计算复杂度。
3 结论
本文提出面向蛋白质功能预测和有向无环图标记结构的多示例多标记学习算法。首先,算法训练一个共享矩阵将高维示例映射到低维子空间;然后,利用SGD优化排序方法初步训练出分类模型;最后,算法考虑了标记间的DAG结果,通过合并多个子节点为超节点,实现对标记的最终预测。本文运用MIMLDAG 算法对多个蛋白质生物学功能预测数据集进行了学习。实验表明,相比于其他类似算法, MIMLDAG拥有更好的预测性能与时间效率。在后续的研究中,拟考虑更多的数据集,进一步扩大算法的应用场景。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
肝博士(2022年3期)2022-06-30
河北农业大学学报(2022年2期)2022-04-26
海外星云(2021年9期)2021-10-14
考试与招生(2021年5期)2021-05-25
实用肿瘤学杂志(2020年4期)2020-12-08
幽默大师(漫话国学)(2020年10期)2020-10-29
娃娃乐园·3-7岁综合智能(2016年6期)2016-09-19
医学综述(2011年12期)2011-12-09
高中生·青春励志(2009年11期)2009-12-03
