
1例Gitelman综合征合并亚临床甲状腺功能减退症患者SLC12A3基因突变位点鉴定及遗传分析
2022-06-06 13:07李敏黄稳莹张甜郭英
山东医药 2022年15期
李敏,黄稳莹 ,张甜,郭英
1 潍坊医学院附属医院内分泌代谢病科,山东潍坊 261000;2 潍坊医学院
Gitelman 综合征(GS)又称家族性低钾低镁血症,是一种罕见的常染色体隐性失盐性肾小管疾病,发病率为1~10/40 000,多于青春期或成年期发病[1]。GS 主要临床特征是低钾血症、代谢性碱中毒、低镁血症、低钙尿症,常累及骨关节、肾脏、心血管、消化和神经系统[2]。GS 有遗传异质性,由SLC12A3 基因突变引起,目前已知的SLC12A3 基因突变超过500 个,该基因编码肾脏远曲小管噻嗪类利尿剂敏感的钠氯共转运蛋白(NCC),GS 诊断金标准是基因检测[3]。研究[4-5]表明,甲状腺激素水平会影响肾脏的生理和发育,慢性肾病和肾小球肾炎等肾脏疾病与甲状腺功能异常密切相关,包括甲状腺激素水平降低和桥本甲状腺炎。GS 合并甲状腺疾病的报道在国内外较少,临床表现缺乏特异性,起病隐匿,易漏诊和误诊。本研究对1 例GS 合并亚临床甲状腺功能减退症患者及家庭成员进行了全基因组测序分析,以鉴别出GS 合并亚临床甲状腺功能减退症的致病基因突变位点。
1 资料与方法
1.1 临床资料 患者女,36 岁,因“间断性全身乏力伴四肢麻木 1 年,加重 20 天”于 2021 年 7 月 30 日入院。1 年前患者无明显诱因出现全身乏力伴四肢麻木,呈间断性发作,每年发作5~6 次,未诊治。20 d 前患者上述症状较前加重,伴憋气,于当地医院查血钾2.2 mmol/L(正常范围3.5~5.3 mmol/L),补钾治疗后症状缓解,复查血钾3.9 mmol/L,患者自行停药。入院前1 周患者再次出现上述症状,查血钾2.9 mmol/L,补钾后症状未改善,遂就诊于本院。患者既往体健,否认高血压病史,无重金属、甘草类制剂、抗精神类药、利尿剂等毒物和药物接触史。体格检查:体温 36.2 ℃,脉搏 90 次/min,呼吸 19次/min,血压 108/66 mmHg(1 mmHg=0.133 kPa),体质量 63 kg,身高 167 cm,BMI 22.5 kg/m2。神志清、精神可,发育正常,营养中等,大小便正常。无突眼及满月脸,甲状腺无肿大。四肢肌力正常、肌张力低。心肺腹未见异常。入院后生化检测结果示血钾2.74 mmol/L,血镁 0.63 mmol/L(正常范围 0.7~1.1 mmol/L),血钙2.38 mmol/L(正常范围2.04~2.71 mmol/L),尿钾84.6 mmol/24 h(正常范围25~100 mmol/24 h),尿钙 0.59 mmol/24 h(正常范围2.5~7.5 mmol/L),立位肾素318.1 mIU/L(正常范围4.4~46.1 mIU/L),卧位肾素117.2 mIU/L(正常范围2.8~39.9 mIU/L),提示患者存在低血钾、低血镁、低尿钙、肾素水平增加。肾上腺CT:左侧肾上腺内侧肢略增粗。胸部CT:右肺纤维结节。心脏超声示:心包积液(微量)。其余垂体—肾上腺轴、性腺轴生化检查、心电图均正常。患者的弟弟35岁,自诉从15岁起活动后出现四肢无力、肌肉痉挛,胸闷憋气症状,血液生化检测曾发现血钾低,血压偏低(具体不详),嗜盐,未进行系统治疗;患者之父患高血压病,患者之母患甲状腺功能亢进症,均无GS 的临床表现。
患者血液生化检测结果:游离甲状腺素(FT4)19.94 pmol/L(正常范围11.97~21.88 pmol/L),游离三碘甲状腺原氨酸(FT3)5.17 pmol/L(正常范围3.1~6.8 pmol/L),促甲状腺激素(TSH)4.97 μIU/L(正常范围0.27~4.2 μIU/mL),促甲状腺受体抗体(TRAb)<0.8 IU/L(正常范围0~1.75 IU/L),抗甲状腺球蛋白抗体(Anti-TG)13.4 IU/mL(正常范围0~115 U/mL),抗甲状腺过氧化物酶抗体(Anti-TPO)10 IU/mL(正常范围0~34 IU/mL),提示患者存在亚临床甲状腺功能减退症。患者之母患甲状腺功能亢进症。在取得家庭成员同意的前提下,采集家庭成员外周血2 mL(EDTA 抗凝),进行致病突变基因位点鉴定。
1.2 GS致病突变基因位点鉴定 在患者及家属同意的前提下进行全外显子组测序检测,抽取患者及父母、弟弟外周血2 mL(EDTA 抗凝血)提取基因组DNA。对基因组DNA 采用IDT The xGen Exome Research Panel v2.0 全外显子捕获芯片,捕获与GS 相关基因的全部编码区序列,进行突变筛查。根据患者突变筛查结果及临床信息,通过OMIM 数据库,MedGen、致病突变数据库、正常人基因组数据库、遗传病数据库等筛选所有与疾病相关的突变基因,根据美国医学遗传学和基因组学会(The American College of Medical Genetics and Genomics,ACMG)对该变异分级解读,筛选等级为致病性的突变位点。对筛选的突变基因位点所在区域进行聚合酶链反应(PCR)扩增,经 ABI3730 测序仪进行 Sanger 测序验证,并经序列分析软件得到验证结果。
2 结果
患者及弟弟SLC12A3 基因存在复合杂合突变c. 1964G>A 及 c. 1456G>A,分别导致 cDNA 第 1964位核苷酸鸟嘌呤(G)突变为腺嘌呤(A),cDNA 第1456 位核苷酸鸟嘌呤(G)突变为腺嘌呤(A),根据ACMG 指南,c. 1964G>A 、c. 1456G>A 均为致病突变,诊断为GS。表型正常家系成员检测到1 处杂合突变,母亲携带c. 1456 G>A 杂合变异,父亲携带c.1964 G>A 杂合变异,均无GS临床表现,为基因突变携带者。家系验证结果(见图1)显示患者及弟弟的突变分别来自于其父母,为杂合突变,符合常染色体隐性遗传。
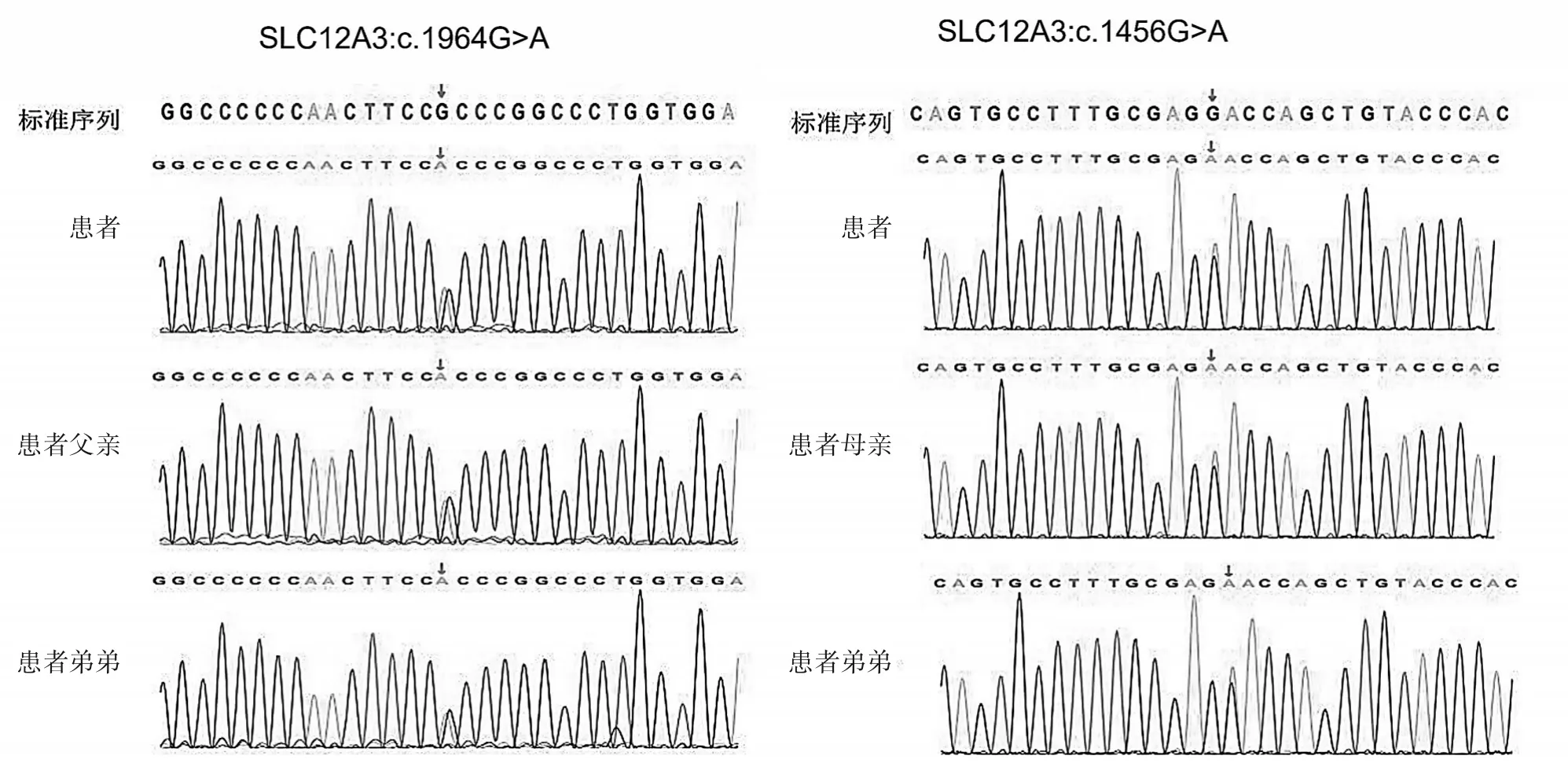
图1 SLC12A3基因c.1964G>A,c.1456G>A变异位点Sanger测序结果
3 讨论
GITELMAN 等[6]在 1966 年首次报道了,GS 临床症状主要是低血钾、低血镁在全身多系统的表现,有疲乏、肌无力、心悸、夜尿增多等,GS 通常合并糖代谢异常、生长发育与骨关节改变、肾小管疾病等慢性并发症,最常合并甲状腺疾病,机制尚不明确[7]。治疗方法主要是钾、镁替代[2]。GS 发病机制为SLC12A3 基因突变导致的 NCC 缺陷[8]。SLC12A3 基因定位于染色体16q13,长约55 kb,含有26 个外显子。目前已发现586 个SLC12A3 基因突变位点,研究发现 T60M、D486N、R913Q 和 R928C 突变在中国最为常见。突变类型包括错义变异、剪切位点变异、无义变异和移码变异,错义变异在GS 中最常见[9]。大多数的GS 患者为复合杂合突变,纯合突变、3 个及 3 个以上的突变也有报道[10]。SLC12A3 基因编码的NCC 是含有1 021 个氨基酸的多肽,有12 个跨膜结构域。NCC 缺陷影响钠和氯在肾远曲小管重吸收:钠重吸收减少,导致Na+/H+交换及Na+/K+交换代偿性增加,引起低钾血症、代谢性碱中毒。氯再吸收减少导致远曲小管膜超极化,进而电压门控顶端膜钙通道开放,钙再吸收增加,引起低钙尿症[11]。JIANG 等[12]推测基因突变不仅影响 NCC,也通过下调瞬时受体电位阳离子通道6(TRPM6)引起低镁血症。GS 的诊断金标准是基因检测。
本例患者的血液生化检测示FT3、FT4 水平正常,TSH 水平升高,诊断为亚临床甲状腺功能减退症,母亲“甲状腺功能亢进症”病史多年,弟弟未行甲状腺功能检测。经Sanger 测序发现患者、弟弟及母亲SLC12A3 基因均存在突变位点c.1456G>A(p.Asp 486 Asn),患者及弟弟发病,携带同一基因突变的母亲无GS 阳性体征,行家系遗传筛查,患者及弟弟c.1456G>A 的突变来源于母亲,故推测SCL12A3基因中c.1456 G>A 突变与甲状腺功能相关。阅读文献发现c.1456 G>A 突变与甲状腺疾病相关性研究报道仅有 1 例。2018 年 LIU 等[13]对 1 例 16 岁男性GS 伴Graves 病(GD)的患者及其三代内的家庭成员进行SLC12A3 基因的遗传分析,发现该名男性患者及其妹妹、父亲、叔父、表姐SLC12A3 基因存在c.1456G>A 突变位点,均合并不同程度的甲状腺疾病(包括TSH 水平升高或甲状腺自身抗体升高),有两个突变位点的家系成员患有更严重的甲状腺功能疾病;其外公及母亲SLC12A3 基因携带(c. 2102-2107delACAAGA)基因突变,未见甲状腺功能异常。在本研究中,SLC12A3 基因携带 c.1456 G>A 突变的患者及母亲均有甲状腺疾病,患者弟弟未行甲状腺功能检测,父亲SLC12A3 基因携带c.1964G>A 突变无甲状腺功能异常,故推测c.1456G>A 突变与甲状腺功能异常相关,但还需进一步进行探究。
通过阅读文献发现,血清镁和甲状腺激素水平密切相关:镁通过激活磷脂酶C-γ1 参与了T 淋巴细胞的活化[14],而自身免疫性甲状腺疾病的发生与T淋巴细胞相关;镁还参与了ATP 的合成,而碘的转运依赖ATP 提供能量,所以镁可以通过影响碘的转运引起甲状腺激素水平的变化[15]。有关镁与甲状腺功能的报道并不少见,MONCAYO 等[15]指出补充镁可以改善甲状腺的形态和功能。CHANDRA 等[16]长期用硫酸镁喂养小鼠后,发现小鼠TSH 随着硫酸镁剂量的增大而升高。KRAVCHENKO 等[17]研究发现,甲状腺肿的患者血清镁的浓度较正常人群低。在本研究中,不排除低镁血症与甲状腺功能存在一定的相关性。
研究[18]表明,甲状腺疾病患者会出现疲乏、肌无力、胸闷憋气等与GS类似的临床症状,当GS患者合并甲状腺疾病时,上述症状可能会加剧,需通过实验室检查、基因检测明确。
GS 是一种遗传性肾小管疾病,SCL12A3 基因突变为主要致病原因,确诊该病的关键是基因检测。我们通过全外显子组测序对一个常染色体隐性遗传的GS 家系进行致病突变分析,发现该家系2 个患者是由SLC12A3 基因c.1964G>A,c.1456G>A 错义突变导致的。SLC12A3 基因第16 号外显子的一个错义突变 c. 1964G>A(p. Arg 655 His),导致 cDNA 第1964 位核苷酸鸟嘌呤突变为腺嘌呤,该突变导致第655 位编码的氨基酸由精氨酸(Arg)突变为组氨酸(His);SLC12A3 基因第 12 号外显子的一个错义突变c.1456G>A(p.Asp 486 Asn),导致cDNA 第1456位核苷酸鸟嘌呤突变为腺嘌呤,该突变导致第486位编码的氨基酸由天冬氨酸(Asp)突变为天冬酰胺(Asn)。该复合杂合突变导致NCC 缺陷,蛋白质功能缺失。
总之,导致该患者发病的致病基因为SLC12A3基因,突变位点为 c. 1964 G>A(p. Arg655His)、c. 1456G>A(p. Asp486Asn);c. 1964G>A 位点的基因突变可能与甲状腺功能相关,还需要进一步的基础研究来阐明GS突变与甲状腺功能之间的关联。
猜你喜欢
种子(2021年3期)2021-04-12
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
百科知识(2015年18期)2015-09-10
医学研究杂志(2015年12期)2015-06-10
