
钠离子层状氧化物材料相变及其对性能的影响*
2022-06-04 06:26:20丁飞翔容晓晖王海波杨佯2胡紫霖2党荣彬陆雅翔胡勇胜2
物理学报 2022年10期
丁飞翔 容晓晖† 王海波 杨佯2) 胡紫霖2) 党荣彬 陆雅翔 胡勇胜2)3)‡
1) (中国科学院物理研究所,北京 100190)
2) (中国科学院大学,材料科学与光电技术学院,北京 100049)
3) (中国科学院物理研究所,怀柔研究部,北京 101400)
钠离子电池近年来在大规模储能领域展现出优异的发展和应用前景.由于钠离子层状过渡金属氧化物正极材料(NaxTMO2)具有比容量高、容易制备、电压可调和成本低的优势,在学术界和产业界得到了广泛的关注与研究.但较大的Na+半径和较强的Na+-Na+静电排斥作用,导致NaxTMO2 具有多种结构类型和复杂的结构转变,以及由此形成了多重结构-性能关系.本文详细介绍了NaxTMO2 的结构类型,综述了在Na+脱出/嵌入过程中引发的结构演变,旨在揭示钠离子层状过渡金属氧化物正极材料结构转变机理及其对电化学性能的影响,最后讨论了现存的挑战并提出了改进策略.
1 引言
对可再生绿色清洁能源(如太阳能、风能、水能等)日益增长的开发应用需求迫使人们不断探索廉价、高效的储能新体系.因为钠资源在地壳和海洋中含量丰富、分布广泛且成本低廉,钠离子电池在大规模电能存储上表现出广阔的应用潜力[1].过去的几十年来,各种正极材料得到研究利用,比如层状氧化物[2-4]、聚阴离子类化合物[5,6]、普鲁士蓝类化合物[7,8]、有机化合物[9-11]等.钠离子层状过渡金属氧化物(NaxTMO2,TM 一般为过渡金属,x≤1)与成功制备的锂离子层状氧化物相似,具有比容量较高、制备简单、压实密度高、电压范围可调等优势,得到了大量研究.尽管钠离子层状氧化物的电化学性能还没有达到与锂离子层状材料相同的水准,但是从性价比的角度综合考虑,其仍是一种具有竞争力的钠离子电池正极材料.钠离子层状过渡金属氧化物与锂离子层状过渡金属氧化物具有较大的差异,通常,Na 比Li 更容易与过渡金属分离形成层状结构,没有互占位的情形.目前仅发现Mn,Co 和Ni 三个元素可以在锂离子层状过渡金属氧化物中提供电荷补偿,而具有电化学活性并可以与钠离子形成层状结构的元素种类相对较多,例如:Ti,V,Cr,Mn,Fe,Co,Ni 和Cu 等元素均可以参与形成钠离子层状氧化物且表现出多种性质[2],尤其是其中廉价的Cu,Mn 和Fe 元素,可以广泛使用.
但是,迄今为止在层状氧化物正极材料中仍有两大挑战亟需解决,一是在较高电压充电态下层状结构很难维持稳定,限制了可以稳定可逆脱出/嵌入的Na+的含量,从而很难兼顾高能量密度和长循环寿命这两大指标;二是一部分性能优异的层状氧化物在初始态呈现出缺钠态,如Na2/3TMO2.缺钠的初始相往往为P2 相结构,虽然该结构的材料在钠离子脱出/嵌入过程中展现出相比于O3 相结构较好的稳定性和钠离子导通性,但是缺钠的特性降低了他们首周的充电容量,尤其是与不含钠离子的负极材料匹配全电池时限制了有效比容量的发挥,因此需要预钠化.本文将详细介绍钠离子层状氧化物材料组成、结构和性能之间的联系,尤其是高电压下的结构转变及其对材料电化学性能的影响,希望能为将来设计和优化新型高能量密度钠离子层状氧化物正极材料提供指导和借鉴.
2 钠离子层状氧化物正极材料简介
2.1 钠离子层状氧化物正极材料常见晶体结构
目前钠离子层状过渡金属氧化物正极材料的通式为NaxTMO2,随着Na+含量的变化形成不同结构,常见的结构有O3,P3,O′3 和P2 相,如图1所示.其中过渡金属位置可以由各种金属离子(如Li,Na,Mg,Ti,V,Cr,Mn,Fe,Co,Ni,Cu,Zn,Sn,Ir,Ru 等)占据,这些金属离子的核外电子排布、氧化态、TM—O 键能差异很大,并随着Na+含量的变化构成了不同结构的层状氧化物正极材料.一般情况过渡金属位的离子与6 个氧形成TMO6的八面体构型,共棱连接组成二维过渡金属层,Na+则占据过渡金属层之间的多面体,构成TMO6层与NaO6层交替分布的层状结构.20 世纪80 年代,Delmas 等[12]提出了这些晶体结构的命名分类方法,其中,O 和P 分别代表着钠离子八面体配位(octahedral)结构和三棱柱(prismatic)配位结构.数字代表着氧离子堆垛的TMO6八面体层(TMO6八面体层通常由上下两种不同的氧层组成,例如:A 和B)最少重复层数,例如O3 为ABCABC···、P3为ABBCCA···、P2 为ABBA···和O2 为ABAC···(通常在电极材料电化学脱钠过程中出现).此外,一般在字母后面用符号“ ′ ”区别材料在原结构基础上晶体对称性降低后的结构(如O′3),这通常是因为部分过渡金属离子具有姜-泰勒效应而引起的晶格扭曲.虽然对称性降低后的结构所属的空间群发生了变化,但仍可以用此命名区分特征结构.当符号“ ′ ”放置在数字后面时,一般用来区分与原始材料空间群相同,但是晶胞参数有较大差别的结构(如O3′).

图1 (a)—(e)常见钠离子层状材料的晶体结构示意图,插图为层状结构中过渡金属和钠离子多面体的连接机理示意图;(f)从垂直于过渡金属层的方向观察六方和单斜结构和晶胞参数的区别和联系Fig.1.(a)—(e) Illustrations of crystal structures relevant to the Na+ layered oxide cathode materials,insets are the face-sharing schemes of TMO6 and NaO6 in the layered structures;(f) the view perpendicular to the layer direction highlighting the relationship between the hexagonal and monoclinic unit cells.
O3 型层状氧化物(图1(a))中的晶格氧以面心立方密堆积(FCC)的形式排列,过渡金属离子和Na+交替分层占据其中的八面体位,并通过共边连接.Na+只有一种晶体学位置3b(0,0,1/2),如表1 所示.以α-NaFeO2为代表,属于六方晶系(Hexagonal),空间群为 Rm.如图2 所示,除了Fe元素以外,3d 过渡金属中的Ti,V,Cr 和Co 也能和Na 形成一元的O3 相氧化物.而一元的NaMnO2和NaNiO2的结构则是O′3 相(图1(b)),空间群为C2/m,属于单斜晶系(Monoclinic).这主要是因为Ni3+(3d7)和Mn3+(3d4)具有较强的姜泰勒效应,过渡金属八面体构型发生畸变(沿着八面体对称的顶点拉伸或者压缩),最终导致材料从六方晶系转变为单斜晶系.在O3 相材料脱钠过程中,随着过渡金属离子的价态变化而变成有姜泰勒效应的离子,如Ni2+/3+,也会形成O′3 相[14].当O3 相中的过渡金属位置由两种或多于两种的离子占据并形成蜂巢有序占位时,如Na[Na1/3Ru2/3]O2(Na2RuO3)[15,16]和Na[Na1/3Ir2/3]O2(Na2IrO3)[17],也会引起晶体的对称性降低,空间群从 Rm变为C2/m.

表1 常见的结构对应的空间群和原子坐标Table 1.The space groups and corresponding atomic positions of reported structures.

图2 各种3d 过渡金属离子在钠离子电池层状氧化物中的特点[13]Fig.2.Comparison of the characteristics of 3d TM used in NIB layered cathode materials[13].
P2 相层状氧化物(图1(e))的Na+含量一般在2/3 mol 左右,钠层中的Na+以三棱柱的配位方式存在,过渡金属离子仍以八面体的配位形式并与钠层交替分布.P2 材料也归属于六方晶系,空间群为P63/mmc.结构中形成两种钠离子位置,一种与过渡金属八面体共边连接(Nae,e 取自edge 的首字母),晶体学位点为2d 位(2/3,1/3,1/4),另一种与过渡金属八面体共面连接(Naf,f 取自face 的首字母),晶体学位点为2b 位(0,0,1/4),如图1(e)所示.其中Naf位的Na+因与过渡金属离子面面相接而具有较高静电排斥力,导致Na+在该位置的占位率明显低于Nae位置(约为Nae位占位率的一半).O2 相(图1(d))通常由P2 相脱钠后转变形成,钠离子八面体与过渡金属八面体分别共面和共边连接,空间群为P63mc.P3 相(图1(c))同样属于六方晶系,空间群为R3m,过渡金属八面体的排列取向与P2 相相比多了一种排布方式,因此钠三棱柱分别与过渡金属八面体共面和共边连接,Na+位置为3a(0,0,~0.17).
2.2 钠离子层状氧化物结构预测
钠离子层状氧化物结构丰富,组成繁多,为基础科学研究和大规模产业化应用提供了巨大的探索空间和应用潜力,同时也带来许多不确定性和困难.因此,如果有一种可以精确而又简单的方法预测电极材料结构,将会加速新型层状氧化物的研发.笔者课题组基于近年来对层状氧化物晶体结构和组成的认识和探索,提出用“阳离子势”这一参数指导设计合成或优化层状氧化物正极材料.“阳离子势”定义如下[18]:

图3 总结了已经报道的P2 和O3 相层状氧化物的阳离子势分布.仔细观察发现阳离子势可以很好地区分两种结构.这主要是因为较大的阳离子势表明过渡金属层有一个较大的电子云分布,使相邻过渡金属层之间的氧氧静电排斥力增加,形成一个较大的Na+层间距,形成P2 相结构.当增加钠层中Na+含量时,将会增加Na+和氧负离子间的静电吸引力,降低过渡金属层间的排斥力,更容易形成O3 相结构.因此,根据典型的P2 和O3 相组成的例子,可以得到区分两相结构的分界线,使得研究人员能够提前预测设计组成的层状堆叠结构.

图3 已报道的P2 相和O3 相中层状氧化物的阳离子势[18]Fig.3.Cationic potential of representative P2-and O3-type Na-ion layered oxides[18].
3 钠离子层状氧化物常见结构演变
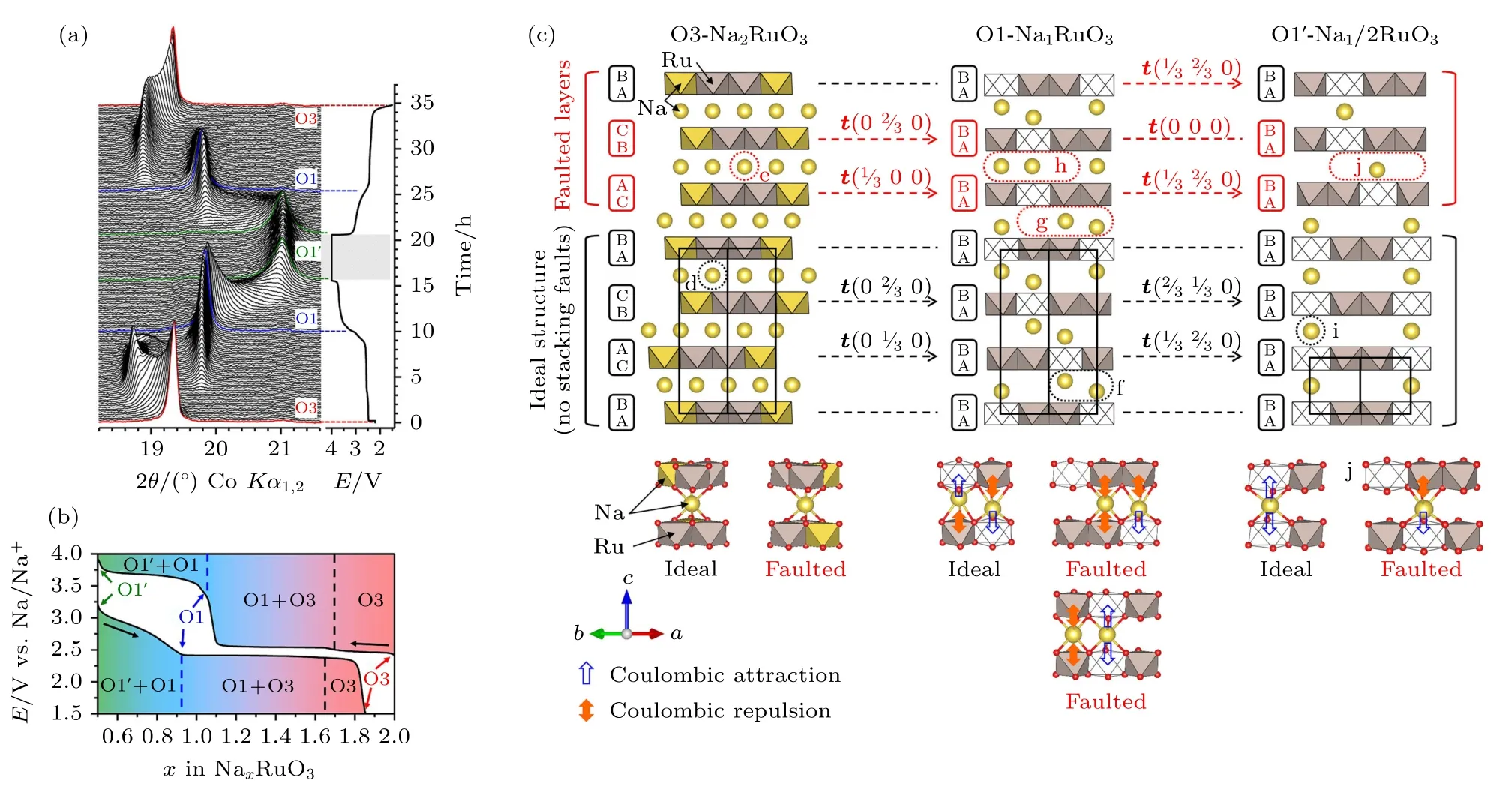
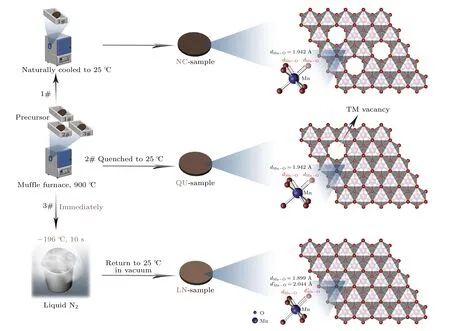
早在20 世纪80 年代,对层状氧化物ATMO2(A 代表Li 和Na)插层化学的发现,大大加速了锂离子电池的商业化.自此,研究者投入大量精力研究理解碱金属离子如何可逆地脱出/嵌入ATMO2材料,因为这直接影响到能否利用其最大理论比容量(LiCoO2约275 mAh/g,NaCoO2约235 mAh/g).目前常见的结论是,在碱金属脱出量较小时,AxTMO2(0.4 图4 当碱金属阳离子和阴离子离子半径分别相同时O和P 型配位环境中的阴离子距离对比[19]Fig.4.Comparison of anion-anion distances in O-and Ptype coordinations assuming the same cation and anion radii[19]. 在A+深度脱出时(0.0 在O3 相材料中,NaNi0.5Mn0.5O2材料具有代表性,该材料相变过程复杂几乎涵盖了O3 相的所有结构转变,适合于分析O3 相发生的相转变类型.图5(a)给出了O3-NaNi0.5Mn0.5O2在不同充电截止电压范围内的充放电曲线.在2.2—3.8 V 内循环时,可以获得125 mAh/g 的可逆比容量,当充电截止电压为4.5 V 时,有185 mAh/g 的可逆比容量,表明有更多Na+可逆地脱出/嵌入该材料的晶体结构.Komaba 等[20]首先通过非原位XRD 表征了O3-NaNi0.5Mn0.5O2在脱钠时的结构转变,如图5(b)所示.首先发现在初始脱钠过程中位于17°(2θ)的O3 相(003)衍射峰劈裂成两个峰,说明有新的结构生成.通过对XRD 特征峰辨别发现新相为单斜的O′3 相,最明显的特征是O3 相(104)衍射峰劈裂形成O′3 相的()和(111)衍射峰,该演变主要是由晶格扭曲导致的晶体对称性下降.当脱出约0.3 mol 的Na+时,发现O′3 相的衍射峰逐渐减弱,P3 相形成,P3 相和O3 相最大的不同是(104)衍射峰显著下降而(105)衍射峰增强.当Na+脱出0.5 mol 时,P3 相发生晶格扭曲转变为单斜的P′3相(类似O3 相向O′3 相的转变),P3 的(105)衍射峰劈裂为P′3 的(201)和(11)衍射峰.在深度脱钠(Na+脱出0.6—1.0 mol)时,XRD 图谱在2θ为12.7,25.3 和37.2°时出现新的衍射峰.虽然作者认为该脱钠相为P3′相,但是此处可能为脱钠态材料的吸水相,主要是由非原位测试过程中极片暴露在环境中导致的.后续Sathiya 等[21]的原位XRD 测试发现不一样的结果,如图5(c)所示,电极材料在4.2 V 左右的充电平台发生了P 相再到O 相的演变(图中区域2).新生成的O 相的(003)衍射峰位于较高角度(约20.3°),说明钠层层间距急剧减小,不利于材料的结构稳定性和循环性能. 图5 (a) NaNi0.5Mn0.5O2 材料在2.2—3.8 和2.2—4.5 V 电压范围内的首周充放电曲线[20];(b) Na1—xNi0.5Mn0.5O2 电极的非原位XRD 图谱[20],星号代表集流体镍网的衍射峰;(c)NaNi0.5Mn0.5O2 材料首圈在C/20C 倍率下充放电过程中的原位XRD 图谱[21]Fig.5.(a) Initial charge-discharge curves of the NaNi0.5Mn0.5O2 cell at a rate of 1/50C (4.8 mA/g) in the voltage ranges of 2.2—3.8 and 2.2—4.5 V versus sodium metal;(b) ex situ XRD patterns of the Na1—xNi0.5Mn0.5O2[20],asterisks show a nickel mesh used as a current collector;(c) in situ XRD patterns of the NaNi0.5Mn0.5O2 material at C/20 rate[21]. 图6 展示了O3 相的晶体结构转变示意图,理想情况下O3 相和O′3 相的晶胞参数满足图6(b)中的矩阵关系,实际上O′3 相晶胞参数a与的比值约为2.89 Å (1 Å=0.1 nm),比b值小1.5%,表明过渡金属层层间出现明显的晶格扭曲.电极材料从O 型到P 型的相变一般在室温恒电流脱钠的条件下.因此,在晶胞中完成相变的唯一可能的方法是依据最小作用原理的层间滑动,而不是层的旋转或TM-O 共价键断裂.因为在层间旋转过程中,远离旋转中心的原子需要移动很长的距离,并伴随着化学键的断裂,而化学键断裂通常在较高的温度下.因此,O3 向P3 的转变主要通过过渡金属层滑移,具体地CA 氧层配位的过渡金属层滑移方向为1/3a+2/3b(其中a,b分别代表着六方晶系空间点阵的基向量),BC 氧层配位的过渡金属层滑移方向为2/3a+1/3b,最终形成钠层三棱柱型的配位形式,晶格氧排布为ABBCCA….同时,晶格扭曲在新的配位形式下得到缓解.但随着脱钠的进行,姜泰勒效应引起的晶格扭曲再次显现出来(形成P′3 相).P3 相和P′3 相的晶胞参数对称性满足图6(c)中的矩阵关系.P′3 相晶胞参数与b值只差仅约为0.5%,说明晶格扭曲程度与O′3 相相比明显较弱. 图6 (a) Na1—xNi0.5Mn0.5O2 电极的晶体结构转化示意图;(b) O3 和O′3 相之间的转化关系;(c) P3 和P′3 相之间的转化关系[20]Fig.6.(a) Schematic illustrations of the crystal structure of Na1—xNi0.5Mn0.5O2;(b) the transforming relationship between O3 and O′3;(c) the transforming relationship between between P3 and P′3[20]. 充电到4.2 V 的电压平台时,层间钠离子不足0.4 mol,为了避免氧离子面面相接,过渡金属层再次滑移形成O 相结构.然而对P3 相有两个滑移方向可以选择,如图7(a)所示,分别为{α}={2/3a+1/3b,—1/3a+1/3b,—1/3a—2/3b} 和{β}={1/3a+2/3b,1/3a—1/3b,—2/3a—1/3b}.滑移方向α倾向于让过渡金属离子与上层毗邻的过渡金属离子(z+1/3)面面接触,而滑移方向β倾向于让过渡金属离子与下层毗邻的过渡金属离子(z-1/3)面面接触.点阵中由于没有垂直于c轴的镜像平面,因此上下不同的滑移方向影响很大.例如在NaNi0.5Mn0.4Ti0.1O2和 NaNi0.4Cu0.1Mn0.4Ti0.1O2体系中在高电压区域就出现明显不同的相转变,分别为P3-O1 和P3-O3′,另外在NaFe1/2Mn1/2O2材料中却发现P3-OP2 的相转变.下文详细介绍较低钠含量时P3 相向O 相转变的各种转化类型. 图7 (a) P3 相过渡金属层可能的滑移方向α 和β;(b) O1 相的晶体结构示意图;(c)通过α 和β 滑移P3 相向O3,O1,OPO13 和O1PO3 相的转变示意 图;(d)通过α 和β 滑移P3 相 向OO13,O1OP3,POO13 和PO1O3 相的转变示意图;(e)通过α 和β 滑移P3 相向OP2 相的转变示意图.晶体结构观察方向为[100],钠位为黄色多面体,过渡金属位于紫色八面体,结构示意图中没有考虑晶胞参数的变化[19]Fig.7.(a) P3 phase TM layer displacement vectors α and β;(b) schematic illustrations of the crystal structure of O1 phase;(c) phase transitions from P3 to O3,O1,OPO13 and O1PO3 via shifting α and β;(d) phase transitions from P3 to OO13,O1OP3,O1OP3 and POO13 via shifting α or β;(e) phase transition from P3 to OP2 via shifting α and β.All structures viewed along [100]direction,all cell parameters changes have been ignored and TM octahedra are shown in purple and all Na sites in yellow[19]. 假设P3 相中有两层过渡金属层可以滑移,总共有4 种组合{β}{β},{α}{α},{α}{β}和{β}{α}分别形成OPO13,O1PO3,O3 和O1 结构,如图7(c)所示.O1,P 和O 分别代表O1,P3 和O3 结构的原子层排布,其中O1 相晶格氧排列方式为ABAB···,钠八面体与毗邻的过渡金属八面体共面连接,钠层层间距减小,如图7(b)所示.假设P 相中仅有一层过渡金属层可以滑移,同样有4 种组合{α}1/3,{α}2/3,{β}1/3和{β}2/3(角标1/3 和2/3 代表滑移层的z位置)形成PO1O3,O1OP3,POO13 和OO1P3结构,如图7(d)所示.由于在六方晶系中OPO13,PO1O3 和O1OP3 以及O1PO3,POO13 和OO1P3是等效的,因此根据最小作用原理仅通过一次的{α}或{β}滑移就可以实现P3 向OPO13 或O1PO3的转变.另外,假如在P3 相两个沿c轴方向的单胞中同时发生4 次滑移,则可以实现P3 到OP2相的转变,如图7(e)所示. 上述相变中最大的不同点在于O1 和O3 相结构的比例,而O3 和O1 相主要由金属离子和配位离子之间键的离子性/共价性决定的.例如在LixTiS2中过渡金属-阴离子(TM-X)更强的共价键性,使其在不同Li+含量中都以O1 结构稳定存在.在LixNiO2体系中,x<0.3 时Ni-O 键较强的共价性使空间效应的作用超过O-O 间静电排斥力的作用,完全脱锂态的NiO2以O1 型结构存在[22].因此可以推断在较低钠含量的充电态下,随着过渡金属-氧键的共价性的增强,稳定的电极材料结构倾向性排序为OP2 O3 型的Na2RuO3材料跟一般的O3 相材料相比,则展现出不同的相变过程[15,23].该材料跟富锂锰基材料Li2MnO3相似,过渡金属层的Na+伴随着氧离子的氧化还原可以可逆地脱出/嵌入,因此显现出不同的结构转变.从图8(a)中的原位XRD图谱可以看出充放电过程中该正极主要经历了O3-Na2RuO3,O1-Na1RuO3和O1′-Na1/2RuO3之间的两个两相转变.在第一个充电平台(2.7 V)发生的是O3-Na2RuO3和O1-Na1RuO3之间的两相转变.O1-Na1RuO3的晶格氧以ABAB…的形式排布,蜂巢有序分布的[Ru2/3□1/3]O2和[Na2/3□1/3]O2(□为空位)层交替排布.在O1 相中钠层的NaO6八面体和过渡金属层共面相接.此时,钠层的钠离子与过渡金属层中的Ru5+和□之间分别存在较强的库仑斥力和库仑吸引力,会导致过渡金属层的滑移更倾向于让钠层的NaO6八面体与过渡金属层的空位八面体相接(图8(c)).最终导致初始O3 相自由的堆叠方式(有堆叠层错)向有序排布转变.但是局域稳定的Na+-□-Na+构型,导致O1 相中仍有层错存在.在第2 个充电平台时,新相O1′-Na1/2RuO3是由O1-Na1RuO3转变形成的,钠层层间距从5.21 Å减小到4.91 Å.此时O1′相的过渡金属层和钠层占位情况为蜂巢有序的[Ru2/3□1/3]O2和[Na1/3□2/3]O2,□-Na+-□-Na+构型进一步平衡静电作用.因此过渡金属层排布更加有序,堆叠层错消失.放电过程则为完全相反的过程.相似的结构转变也在Na2IrO3[17]中存在. 图8 (a) Na2RuO3 的原位XRD 图谱和充放电曲线;(b)根据原位实验确定的随钠含量变化的相图;(c)库仑力对Na2—xRuO3 材料自有序过程的机理演示[23]Fig.8.(a) XRD patterns tested in situ during the first cycle of Na2RuO3 with the corresponding cycling curve;(b) phase diagram as determined from the in situ experiment as a function of the sodium content;(c) coulombic forces and resultant self-ordering in Na2—xRuO3[23]. 除了过渡金属层的整体滑移之外,金属离子在充电过程中也有可能迁移到相邻的碱金属层中.因此层状材料在充电态下的结构稳定性有时与金属离子的迁移密切相关,并有可能影响到电化学性能.特别是,脱出/嵌入Na+时不可逆的过渡金属离子迁移可能导致材料的热稳定性下降,进一步导致容量和电压迅速衰减.在锂离子层状材料中,由于Li+和金属离子的离子半径接近,阳离子迁移行为十分普遍.钠离子的离子半径较大,因此充电时金属离子的迁移主要发生在O3 型层状氧化物中,例如O3-NaTiO2[24],O3-NaVO2[25],O3-NaCrO2[26]和 O3-NaFeO2[27]. 图9(a)和9(b)给出了NaFeO2和NaCrO2电极在不同电压范围内的充放电曲线.可以发现随着充电截止电压的提升,材料的放电比容量却在下降.当充电截止电压设置到4.5 V 时,两个材料的放电比容量几乎接近于0,表示结构可逆性遭到严重的破坏.Komaba 等[26]进一步结合原位XRD 表征研究发现NaCrO2电极在充电初始过程中主要经历O3-O′3-P′3 的相变,当充电到3.8 V 的平台时(将近脱出0.7 mol 的钠离子),出现P 相向O相的相变,但形成的O 相结构中铬离子不是完全占据过渡金属八面体的位置.这表示铬离子发生了一定的迁移,初步推测铬离子可能占据了钠层的四面体位或八面体位.图9(c)给出了铬离子占据不同位置的XRD 模拟图谱,发现充电末态的XRD图谱与铬离子同时占据钠层的八面体位置和四面体的拟合XRD 图谱相一致.结合DFT 计算结果,研究人员提出当Na+脱出超过0.5 mol 时,P 相结构开始变得不稳定并通过过渡金属层滑移形成O3′型结构.进一步铬离子会从过渡金属八面体位通过钠层的四面体位迁移到钠层中的八面体位,如图9(e)所示.由于该过程不可逆,导致可逆容量的损失.在NaFeO2体系中,研究人员[28]发现当Na+大概脱出0.5 mol 时铁离子开始迁移,如果迁移量较少(例如充电到3.6 V),Na+可以回嵌并促使铁离子迁移回初始位置.但是当铁离子迁移较多超过一定的门槛(如充电到4.0 V),材料结构更倾向于形成“X”相(未定义的相结构),钠离子脱嵌可逆性降低且极化增加,大部分铁离子也不能回到初始位置,从而造成容量的急速衰减. 图9 (a) O3-NaFeO2 和(b) O3-NaCrO2 电极在不同充电截止电压的充放电曲线[26,27];(c)充电至高电压时Na1—xCrO2 材料模拟和测试的XRD 图谱;(d) NaxFeO2 材料在钠离子脱出过程的相图和铁迁移示意图[28];(e)脱钠过程中过渡金属离子迁移机理示意图[29]Fig.9.Charge-discharge curves of (a) NaFeO2 and (b) NaCrO2 cathode[26,27]; (c) simulated and observed XRD patterns of Na1—xCrO2 cathode charged to high voltage;(d) scheme of phase evolution and iron migration upon sodium extraction in NaxFeO2[28];(e) a proposed mechanism of Men+ (Metal ion) migration process on the desodiated process[29]. 图10 总结了O3 型钠离子层状正极材料在脱嵌钠过程中常见的充放电曲线,以及对应的结构转变行为.其中,4.0 V 以下电压内出现的六方晶系向单斜晶系的转变可以通过过渡金属替代或掺杂消除.当充电到4.0 V 以上时,P 相向O1 相的转变和过渡金属离子迁移也可以通过过渡金属替代得到一定的缓解,形成晶胞参数变化较小的OPn和O3′相.但是该区域的结构转变对材料电化学性能的影响远高于低电压下结构转变对材料综合性能的影响,并决定着该类型材料较高的能量密度是否能得到高效利用,因此需要深入的研究. 图10 充放电过程中O3 型材料的结构演变汇总[30]Fig.10.Summary of structure evolution for O3-type materials during the charge-discharge process[30]. 钠离子P2 相层状电极材料,在充放电过程中经历的相变过程主要有:1)P2 相范围内的Na+/空位有序无序转变;2)深度脱钠后P2 相向O2,OP4或者“Z”相的转变;3)额外嵌钠时P2 相向P′2 相的转变.下面我们主要以P2-Na2/3Ni1/3Mn2/3O2为例,介绍相变的详细过程. 图11(a)展示了P2-Na2/3Ni1/3Mn2/3O2电极材料在2.0—4.5 V 电压区间内的充放电曲线.其放电比容量约160 mAh/g,接近理论比容量173 mAh/g,证明结构中的Na+几乎可以实现全部的可逆脱出/嵌入.与O3 相相比,P2 相材料明显具有更高的工作电压,这主要是因为缺钠和晶格氧不同的构型造成的.笔者课题组在P2-Na0.68Cu0.34Mn0.66O2中首次发现了Cu3+/Cu2+氧化还原电对在钠离子氧化物中具有电化学活性[31],为后续的材料设计和组成优化提供了更多可能.Lu 等[32]首先通过原位XRD 测试研究了P2-Na2/3Ni1/3Mn2/3O2在脱钠过程中的结构转变,如图11(b)所示.从XRD 图谱的改变,可以发现在初始脱钠过程(充电到3.9 V)中P2 相的(00l)和(10l)衍射峰先向低角度偏移,而(110)和(112)衍射峰向高角度偏移,表明随着Na+的脱出晶胞参数c增大a减小,结构仍保持P2 相.虽然XRD 图谱没有发现新的结构出现,但是充放电曲线两个明显的平台表明可能存在两相转变.后续深入的研究也确实发现在Na+含量为2/3,1/2 和1/3 时存在不同的Na+和空位有序排布的结构,后面会详细介绍.当Na+脱出约1/3 时,电压曲线到达4.2 V 左右的平台,位于21°和70°左右的新的衍射峰(002′)和(112′)出现,说明有新的相形成.同O3 相高电压区域的结构转变类似,P2 相在高电压态也发生了P 相向O 相的转变,并伴随着巨大的体积收缩(约23%),不同的是新相的结构为O2 结构.较大的体积变化降低电极材料的电子电导和结构稳定性,导致材料的循环性能严重下降. 图11 (a) P2-Na2/3Ni1/3Mn2/3O2 的充放电曲线;(b) P2-Na2/3Ni1/3Mn2/3O2 首周充电过程中的原位XRD 图谱[32]Fig.11.(a) Charge-discharge curve of P2-Na2/3Ni1/3Mn2/3O2;(b) XRD patterns measured during the first charge of the P2-Na2/3Ni1/3Mn2/3O2 cathode in situ cell[32]. 针对3.4 V 和3.6 V 两个电压平台产生的原因,Meng 等[33]研究发现在Na+含量为2/3,1/2和1/3 时,钠层存在钠离子/空位有序,从而形成特定钠含量下稳定存在的相.这主要是层内Na+-Na+的排斥力和层间Na+-TMn+的排斥力(主要来自Naf位)相互作用的结果,如图12 所示.在初始态即钠含量为2/3 时,Naf位的Na+(约占据总Na+含量的1/3)间距为2ahex并以“large zigzag”形式分布.Na+含量为1/2 时,Naf和Nae位的Na+以1∶2的比例呈队列状排布.Na+含量为1/3 时,两种占位的Na+以1∶1 的比例呈队列状分层交替排布. 图12 (a) P2-Na2/3Ni1/3Mn2/3O2 的典型充电曲线;(b) P2-NaδNi1/3Mn2/3O2 层内三棱柱位置钠离子/空位有序排布示意图(蓝色球代表占据Nae 位钠离子,粉色球代表占据Naf 位钠离子)[33]Fig.12.(a) Typical charge profiles of P2-Na2/3Ni1/3Mn2/3O2;(b) in-plane Na+/vacancy orderings of P2-NaδNi1/3Mn2/3O2 in the triangular lattice (blue balls:Na-ions on Nae sites,pink balls:Na-ions on Naf sites) [33]. Wang 等[34]通过对P2-Na2/3Ni1/3Mn2/3O2材料进行Ti4+替代,合成组成为P2-Na2/3Ni1/3Mn1/3Ti1/3O2的材料,研究发现:Ti4+替代一方面可以有效抑制Na+/空位有序,使替代后的材料在较宽的电压区间内仅发生P2 单相固溶反应(见图13);另一方面降低材料中两种不同占位的Na+的占位能,使Na+具有更好的扩散性能.替代后的材料因此展现出平滑的充放电曲线,并具有出色的循环性能和倍率性能. 图13 (a) P2-Na2/3Ni1/3Mn2/3O2 和(b) P2-Na2/3Ni1/3Mn1/3Ti1/3O2 电极在2.5—4.3 V 电压范围内首周充电过程中的原位XRD 图谱[34]Fig.13.In situ XRD patterns of (a) P2-Na2/3Ni1/3Mn2/3O2 and (b) P2-Na2/3Ni1/3Mn1/3Ti1/3O2 electrodes during the first charge between 2.5 and 4.3 V[34]. 由于P2 相材料本身处于缺钠状态,在首周放电过程中如果继续放电到较低电压下,如1.5 V时,会有额外的Na+(一般来自钠金属负极)嵌入到晶体结构内.图14 显示在1.5—4.0 V 电压范围内P2-Na2/3Ni1/3Mn2/3O2可以实现151 mAh/ g 的放电比容量,1.75 V 左右的电压平台贡献了将近70 mAh/g 的放电比容量,主要是由Mn4+还原为Mn3+提供电荷补偿[35].大量具有姜泰勒效应的Mn3+离子,导致过渡金属八面体畸变,形成晶格扭曲的P′2 相结构(空间群为Cmcm).同样的现象也会出现在P2-Na2/3MnO2[36]材料中,Mn4+/3+氧化还原中心往往具有较低的工作电压,会显著降低体系的能量密度,而且姜泰勒效应会破坏层状结构的稳定性并降低循环性能,因此该氧化还原电对的应用仍然需要进一步合理设计与优化. 图14 P2-Na2/3Ni1/3Mn2/3O2 电极在1.5—4.0 V 电压范围内的首周充放电曲线[35]Fig.14.Initial charge-discharge curves of P2-Na2/3Ni1/3 Mn2/3O2 in the voltage of 1.5—4.0 V[35]. 图15 展示了P2 相在高电压区域向O 相转变的结构示意图.在P2 相中临近的过渡金属层之间面面相接,所以P2 相过渡金属层仅有一个可滑移的等效滑移方向,如图15(a)所示,为{γ}={2/3a+1/3b,—1/3a+1/3b,—1/3a—2/3b,1/3a+2/3b,1/3a—1/3b,—2/3a—1/3b}.P2 相单胞中仅有两层过渡金属层,仅需一个过渡金属层滑移,就可以实现向O2 相的转变,如图15(b)所示.但是,当沿c轴方向的两个单胞中相邻的过渡金属层同时协同发生滑移时,则会形成OP4 结构(P2 相和O2 相交替排布),如图15(c)所示.当更多的过渡金属层协同参与滑移时,还会产生更加复杂的P2 和O2 共生相,一般称作“Z”相.研究表明最终形成Z 相或者OP4 相的材料,体相的Na+含量相对较大,充电比容量较小,结构可逆性相对较好.说明体相Na+的含量对结构的稳定性至关重要,因此一方面可以通过提高初始材料的Na+含量(即设计高钠含量的P2 相),使其体相Na+含量在充电末态保持相同的情况下提供更多稳定的放电比容量;另一方面通过优化组成设计,使其在高脱钠情况下的结构稳定性能够得到有效保持.例如,笔者课题组设计了一种P2-Na0.72Li0.24Mn0.76O2材料,通过原位和非原位XRD 表征技术发现该材料在4.5 V 的充电态仍保持P2 结构,透射电子显微镜表征显示仅在材料颗粒的边缘处发现少量O2 相结构[37].这主要是因为充电过程中O2—被氧化成O—等,能够降低过渡金属层之间的静电排斥,使充电态结构维持在P2 相.但是一些其他氧离子参与电荷补偿的材料,如Na2/3ZnxMn1—xO2[38],Na2/3MgxMn1—xO2[39]等,还是有一定程度的P2 向OP4,Z 或者O2 相的转变. 图15 (a) P2 相过渡金属层可能滑移的方向γ;(b)通过γ 滑移P2 相向O2 相的转变示意 图;(c)通过γ 滑移P2 相向OP4 相的转变示意图.晶体结构观察方向为[100],钠位为黄色多面体,过渡金属位于紫色八面体,结构示意图中没有考虑晶胞参数的变化[19]Fig.15.(a) P2 phase TM layer displacement vectors γ;(b) phase transitions from P2 to O2 via shifting γ;(c) phase transitions from P2 o OP4 via shifting γ.All structures viewed along [100] direction,all cell parameters changes have been ignored and TM octahedra are shown in purple and all Na sites in yellow[19]. 近日,笔者课题组[40]结合球差电镜及第一性原理计算等方法,通过比较两种带状有序(Li+和过渡金属离子有序排布)的层状氧化物正极P2-和P3-Na0.6Li0.2Mn0.8O2(NLMO),确定了拓扑保护对提高晶格氧氧化还原可逆性的关键作用.在此项研究中,首先结合球差电镜及第一性原理计算确定了NLMO 带状过渡金属层的堆积序列,即一维拓扑结构(ODT),原始的P2-和P3-NLMO 中分别为-α-β-堆积和-α-γ-堆积.电化学和结构分析证实,在P3-NLMO 中,-α-γ-堆积在钠离子脱嵌过程中保持不变,其稳定的拓扑特征为可逆氧离子的氧化还原提供了拓扑保护,而P2-NLMO 中-α-β-堆积的拓扑特征则不能稳定保持,在循环过程中逐渐从-α-β-堆积演变为-α-γ-堆积,而-α-γ-模型容纳更少的钠离子,导致容量衰减(见图16).我们提出利用一维拓扑序来重新定义P3-NLMO 结构,对应的拓扑序为[1 3 5 ··· 2q+1],而P2-NLMO 为[1 2 3 ··· q].区别于传统相(O 型或P 型)定义,拓扑序作为层状正极的一个新序参量,可以用来描述不均匀过渡金属层之间的相互作用.在本工作中,P3-NLMO所具有的奇数型拓扑序更有利于维持结构的稳定性,从而提升氧离子的氧化还原的可逆性.P3-NLMO正极在钠半电池中,在电压范围为2.0—4.8 V 和电流密度10 mA/g 的条件下,在第2 个循环中提供了约240 mAh/g 的放电容量,在30 个循环后显示出98%的容量保持率;而P2-NLMO 容量为183 mAh/g,30 圈后容量保持率仅为60%.这项工作为开发高能量、低成本、环境可持续和安全的正极材料提供了强有力的指导. 图16 (a) (b) P2-和P3-Na0.6Li0.2Mn0.8O2 正极材料钠离子脱嵌过程中的拓扑保护机制[40]Fig.16.(a)(b) Topological protection mechanism during Na-ion deintercalation of P2-and P3-Na0.6Li0.2Mn0.8O2[40]. 图17 总结了P2 型钠离子层状正极在脱嵌Na+过程中常见的充放电曲线,以及对应的结构转变行为.与O3 相不同的是,在4.0 V 以下电压范围没有O 相和P 相之间的转变,但有可能会出现Na+/空位有序的结构转变和额外Na+嵌入时P2 相向P′2 相的转变.当充电到4.0 V 以上时,P2 相向O2 或者OP4 相的转变同样对材料的结构稳定性和电化学性能带来不利的影响. 图17 充放电过程中P2 型材料的结构演变汇总[30]Fig.17.Summary of structure evolution for P2-type materials during the charge-discharge process[30]. P2 型钛基钠离子层状材料(NaxTMyTi1—yO2)因其中的Ti4+/3+具有合适的氧化还原电位(约0.7 V vs.Na+/Na)和结构中富含的Na+空位,适用于钠离子电池负极材料[41].该体系材料的结构演变行为与P2 型锰基正极材料表现截然不同,十分值得探究.为了获得更多的比容量,该类材料的钠含量通常在2/3 mol 左右,可以实现大约1/3 mol钠离子的可逆嵌入/脱出,对应的理论比容量在100—120 mAh/g 范围内.通常结构中钛离子以正四价的形式存在,在空气中具有较好的稳定性,因此TM 位需要低价态阳离子掺杂维持电荷平衡.目前已经报道的组成有Na0.66Li0.28Ti0.72O2[41],Na0.6Cr0.6Ti0.4O2[42],Na2/3Ni1/3Ti2/3O2[43],Na2/3Co1/3Ti2/3O2[44],Na2/3Ni1/6Co1/6Ti2/3O2[45],Na2/3Mg1/6Ni1/6Ti2/3O2[46]和Na0.65Li0.13Mg0.13Ti0.74O2[47].这些材料在用作负极时均表现出较好的循环稳定性,研究证实该类型材料在充放电过程中(Na+含量在0.60—1.0 之间)发生的都是固溶反应,没有相变.如图18 所示,Na0.66Li0.28Ti0.72O2和Na0.6Cr0.6Ti0.4O2的充放电曲线为光滑的斜线,没有平台区域出现,原位XRD 图谱没有新的衍射峰出现,仅观察到衍射峰的偏移(由Na+含量和过渡金属离子半径变化导致).相比于在富Na+含量时明显向P′2 相转变的P2 型锰基正极材料,P2 型钛基材料富钠状态下大量的Ti3+(3d1)的电子构型没有明显的姜泰勒效应,不会造成过渡金属八面体配位的扭曲,稳定了结构,避免了相转变的发生. 图18 (a) Na0.66Li0.22Ti0.78O2 电极在0.1 C 倍率下0.4—2.5 V 电压范围内的充放电曲线;(b) Na0.66Li0.22Ti0.78O2 电极在C/7 倍率下首周充放电过程中的原位XRD 图谱[41];(c) Na0.6Cr0.6Ti0.4O2 电极在0.1 C 倍率下0.5—2.5 V 电压范围内的首周充放电曲线[42];(d) Na0.6Cr0.6Ti0.4O2 电极在C/5 倍率下首周充放电过程中的原位XRD 图谱[42]Fig.18.(a) The discharge-charge curves of Na0.66Li0.22Ti0.78O2 at a current rate of 0.1 C (10.6 mA/g) in the voltage range of 0.4—2.5 V;(b) in situ XRD patterns collected during the first discharge-charge of the Na0.66Li0.22Ti0.78O2 electrode under a current rate of C/7[41];(c) the first discharge-charge curve of Na0.6Cr0.6Ti0.4O2 in the voltage range of 0.5—2.5 V;(d) in situ XRD patterns collected during the first discharge-charge of the Na0.6Cr0.6Ti0.4O2 electrode under a current rate of C/5[42].. 上文介绍了钠离子层状材料的结构类型和相转变,下文将针对不同元素组成(包括掺杂改性、元素替代等)、不同合成方法、不同结构类型和包覆改性策略对层状材料电化学性能的影响. 一元钠离子层状正极材料如O3-NaCrO2[48],O′3-NaMnO2[49,50],P2-Na0.67MnO2[36],O3-NaFeO2[51],O3-NaCoO2[52],P2-Na0.67CoO2,O′3-NaNiO2[48,53,54]等,普遍存在相变复杂,结构不稳定或者过渡金属离子迁移的问题,很难提供可靠的电化学性能.但是在这些材料的基础上通过其他元素的替代或者掺杂可以显著提高材料的结构稳定性以减少相变,甚至抑制过渡金属离子迁移. Komaba 等[20]通过用0.5 个Ni2+和0.5 个Mn4+替代O′3-NaNiO2中的Ni3+设计合成了六方晶系的O3-NaNi0.5Mn0.5O2,不仅提高了放电比容量,还在一定程度上减少了相变的发生.Kim 等[55]通过更加便宜的铁离子替代合成了O3-NaNi1/3Fe1/3Mn1/3O2,与硬碳匹配组装的全电池获得了较好的循环性能.笔者团队基于Cu3+/Cu2+氧化还原电对的电化学活性,优化设计合成了O3-NaCu1/9Ni2/9Fe1/3Mn1/3O2正极材料,表现出更加优异的电化学性能[56].随后马紫峰课题组[57]对NaNi1/3Fe1/3Mn1/3O2材料进行了更加详细的研究,发现该材料在4.0 V 以内循环时仅发生可逆的O3-P3 相转变,如图19 所示.当充电到4.3 V 时,一个新的单斜O 相形成了(文中定义为O3′相),在放电过程则经历了不可逆的O3′-P3′-O3 相的转变.尽管如此,该材料仍展现出了在较宽电压范围内工作的可行性.111 组成的镍铁锰体系同锂电镍钴锰三元组分类似,但是铁离子在钠电层状氧化物的作用还不清楚.Yuan 等[58]合成了一系列组成的O3-NaFex(Ni0.5Mn0.5)1—xO2(x=0,0.1,0.2,0.3,0.4 和1)的材料,研究发现铁离子可以有效减弱钠离子/空位有序,尤其是铁含量为0.2的材料在4.0 V 的截止电压下可以实现131 mAh/g的放电比容量,循 环30 周后仍有95%的容量保持率,10C 倍率下能够保持首周低倍率下64%的可逆比容量.非原位XRD 测试结果表明当充电到4.3 V 时,优化后的材料可以形成更加稳定的OP2相,一定程度上缓解了高电压材料的结构坍塌[59]. 图19 O3-Nax[Ni0.33Fe0.33Mn0.33]O2 在(a) 2.0—4.0 V 和(b) 2.0—4.3 V 之间同步辐射原位XRD 测试结果以及充放电曲线[57]Fig.19.In situ XRD patterns tested during cycling of NaxNi1/3Fe1/3Mn1/3O2 electrode in the voltage range of (a) 2.0—4.0 V and (b)2.0—4.3 V[57]. Ti4+也具有资源丰富和价格便宜的优点,当对O3-NaNi0.5Mn0.5O2替代时也展现出平滑充放电曲线的作用,证明发生的是可逆的O3-P3 相转变[60,61].O3-NaNi0.5Mn0.2Ti0.3O2在4.0 V 的截止电压下可以实现135 mAh/g 的放电比容量,1C 下循环200 周后仍有85%的容量保持率[61].采用非过渡金属Sn4+替代也具有诸多优势,比如:1)Sn4+比Mn4+具有更大的离子半径,可能对稳定O3 结构产生不错的效果;2)Sn4+4d 轨道与氧的相互作用减少了轨道重叠,使电子局域更加稳定地存在过渡金属层,有利于其他金属离子与氧形成更强的离子键,提高体系的氧化还原电位.Tarascon 等[21]合成了一系列的Sn 替代的NaNi0.5Mn0.5—ySnyO2(y=0—0.5)材料,随着Sn 替代量的增加,通过固相法合成的材料中NiO 杂相逐渐减少,说明Sn4+(离子半径更接近Ni2+)更倾向于与Ni2+形成固溶的O3相.结构和电化学测试发现Sn4+替代增加了材料的平均充放电电压,抑制结构转变.文中还考虑了其他d 轨道电子全部占满或者全空的金属离子(如Ge4+(3d10),Ti4+(3d0)和Zr4+(4d10))对氧的电荷局域化和电荷密度的影响(即对材料氧化还原电压的影响).通过计算发现NaNi0.5Mn0.5O2材料中Ni-O 键的离子性提升最低,具体平均电压高低排序为:NaNi0.5Zr0.5O2> NaNi0.5Ge0.5O2≈ NaNi0.5Sn0.5O2> NaNi0.5Ti0.5O2> NaNi0.5Mn0.5O2.值得注意的是,替代元素的选取不仅要考虑对氧化还原电压的影响,还要考虑对结构稳定性的影响,因此还需要进一步优化探索. Cu2+和Ti4+同时对NaNi0.5Mn0.5O2中的Ni2+和Mn4+替代取得了更加优异的综合性能.Yao 等[62]通过Cu/Ti 共替代合成NaNi0.45Cu0.05Mn0.4Ti0.1O2正极材料,空气稳定性测试结果表明与NaNi0.5Mn0.5O2正极材料相比有近20 倍的增长,即使浸泡在水中仍能保留其初始结构,而NaNi0.5Mn0.5O2材料的结构遭到完全破坏.另外,由于抑制了循环过程中的电荷有序化和复杂的相变,替代后样品的电化学性能也得到了明显的改善.Wang 等[63]则更多地关注了Cu/Ti 共替代合成的NaNi0.4Cu0.1Mn0.4Ti0.1O2和仅有Ti4+替代的NaNi0.5Mn0.4Ti0.1O2正极在高电压下相变的不同.原位XRD 表征结果如图20 所示,NaNi0.5Mn0.4Ti0.1O2正极在充到高电压时六方晶系16°左右的衍射峰逐渐消失并在20°左右形成新的衍射峰,说明Na+层间距大幅度减小,新形成的结构类似O1 结构.而NaNi0.4Cu0.1Mn0.4Ti0.1O2正极在充到高电压态时仅在17°左右产生新的衍射峰,根据前面的描述可以定义为OP2 相.两者高电压相的晶胞沿c轴方向的缩聚分别为17%和3%,因此在较宽电压区间内循环时,Cu/Ti 共掺的样品拥有较高比容量,同时也获得了更加优异的循环性能. 图20 (a) NaNi0.5Mn0.4Ti0.1O2 和(b) NaNi0.4Cu0.1Mn0.4Ti0.1O2 电极在2.0—4.5 V 电压范围内的原位XRD 图谱[63]Fig.20.In situ XRD patterns of (a) NaNi0.5Mn0.4Ti0.1O2 and (b) NaNi0.4Cu0.1Mn0.4Ti0.1O2 electrode in the voltage range of 2.0—4.5 V[63]. 随后该课题组[64]研究了Zn2+替代Cu2+对材料高电压结构的影响.原位XRD 图谱显示当充电到4.5 V 以后继续恒压充电时,NaNi0.4Zn0.1Mn0.4Ti0.1O2正极材料仍保持OP2 相,因此锌掺杂的样品也获得了较好的电化学性能.这表明通过合适的元素替代减少相转变,尤其是高电压区域的相变,可以使钠离子层状材料发挥出较高的容量.Kubota等[65]通过Mg 和Ti 共掺杂制备了NaNi4/9Mg1/18Mn1/3Ti1/6O2正极材料,该材料在高电压区间转变为类OP2 相的缓冲相,明显抑制了材料晶胞参数的严重衰减,从而提高其结构和长循环稳定性. 高熵氧化物(HEO)作为一种新型化合物,因其具有独特的特性(通常是一些仅具有一种或几种主要元素的常规材料而无法实现的)而广受科学界关注[66,67].HEO 代表可以结晶为单相的多元素金属氧化物系统,其中不同的系统可以处于不同的晶体结构中,包括岩盐、尖晶石和钙钛矿结构.一般晶体结构中,具有5 个或更多主要元素在HEO 中共享相同的原子位点,可以形成稳定的固溶体状态.由于这些材料的组成极其复杂,它们通常表现出优异的性能,例如高断裂韧性,高强度,良好的高温/低温性能,良好的储能性能等. 作者课题组通过高温固相反应法,成功合成了高熵构型单相O3 型的NaNi0.12Cu0.12Mg0.12Fe0.15Co0.15Mn0.1Ti0.1Sn0.1Sb0.04O2钠离子电池正极材料.通过原位XRD 测试解析其充放电过程中的结构信息发现:充电过程前期空间群 Rm的O3 相先转变为相同空间群晶胞参数略微不同的O3′相,脱出0.32 mol Na+后转变为空间群为R3 m 的P3相(充电截止电压为3.9 V),放电过程P3 相可逆地转变为O3′相,并没有回到初始态的O3 相.第2 周的原位XRD 测试结果表明充放电过程为可逆的O3′⇌ P3转变.值得注意的是,通过对比放电过程中P 和O 相结构内对应的放电比容量发现:O3′相下拥有60%的放电比容量,远高于目前文献中报道的其他O3 型层状正极材料.文中提出高熵材料中多重组分的过渡金属离子可以调节钠离子嵌入/脱出过程中的局部结构,从而使相转变得以延迟并高度可逆(如图21(a)所示).因此该材料具有优异的倍率和循环性能,5C 倍率下仍拥有0.1C倍率测试条件下80%的放电比容量,3C 倍率下循环500 周容量保持率83%.该工作提出的高熵构型的概念,为进一步提高钠离子电池能量密度提供了新的思路. 为了开发具有更高容量和更长循环寿命的钠离子电池正极材料,设计高镍的O3-NaTMO2材料有希望成为具有前景的路径.这一策略与高镍锂电正极材料相似,目前NCM(镍钴锰)材料体系中的镍的摩尔量已经达到80%以上,开发不含钴离子的高镍正极也是锂离子电池的研发目标,但是在减小钴离子的物质的量时遭遇了技术瓶颈(如增加的锂镍互占位、降低的热稳定性等[70]),导致材料成本很难下降.然而,在钠离子电池层状过渡金属氧化物中,情况有所不同:1)Ni2+和Na+具有较大差异的离子半径,在层状氧化物中不会存在Ni 和Na 互占位情况,在层状结构中不存在Ni2+和Na+无序排布的问题,这为合成性能优异的无钴高镍钠电正极材料提供了可能;2)在锂电体系中不能变价提供电荷补偿的过渡金属在钠电体系表现出了电化学活性(如Fe,Cu 等),这为钠电正极材料的开发提供了更多选择. 笔者课题组[69]采用低成本的Fe3+替代Co3+合成了一系列O3 相的高镍材料NaNi0.6Fe0.25Mn0.15O2,NaNi0.7Fe0.2Mn0.1O2和NaNi0.8Fe0.15Mn0.05O2.通过初步电化学评估证实组成为Na[Ni0.60Fe0.25Mn0.15]O2的材料具有较好的综合电化学性能,在2.0—4.2 V 的电压范围内表现出190 mA·h/g 的可逆比容量.结合原位XRD 和非原位XAS 测试分析研究了该材料的储钠机理,结果表明在钠离子脱出/嵌入过程中镍和铁离子参与电荷补偿,同时发生O3-O′3-P3-O3′的可逆相转变过程,如图21 所示.值得一提的是,进一步的非原位XRD,GITT和TEM 测试证实高电压下的O3′相表现出较差的Na+扩散动力学特性和结构热力学稳定性,导致该材料在较高截止电压下具有较差的倍率和循环性能.因此还需要进一步通过表面包覆、痕量掺杂和电解液优化进一步提升其循环稳定性. 图21 (a)高熵构型稳定O3 结构的机理阐释[68];(b) NaNi0.6Fe0.25Mn0.15O2 材料在2.0—4.2 V 电压范围内的原位XRD 图谱和(c)结构演变示意图[69]Fig.21.(a) Possible mechanism of high-entropy composition in facilitating layered O3-type structure[68];in situ XRD patterns of (b)NaNi0.6Fe0.25Mn0.15O2 electrode in the voltage range of 2.0—4.2 V and (c) schematic of structural evolution[69]. 钠离子层状过渡金属氧化物通常是通过简单高温固相法合成的,合成温度、烧结时间、煅烧气氛和降温速度等都对材料的理化性质起到至关重要的影响.因此在设计元素组成的过程中,对合成条件的优化也十分重要. 通常,层状氧化物中的起始钠含量处于较高水平时(通常大于0.8),该材料为O3 相,起始钠含量处于较少水平时(通常位于0.6—0.8 之间),材料为P2 相.图22 所示为Na2/3Fe2/3Mn1/3O2组成在不同合成条件下制备的纯P2 和O3 相的结构、形貌和电化学测 试.P2 相是以Na2CO3,Fe2O3和Mn2O3为原料,经过球磨混合均匀后,最后在氧气气氛1000 ℃下烧结12 h 合成.而O3 相是以NaNO3,Fe(OH)3和Mn(OH)2为原料,经过球磨混合均匀后,最后分别在氧气气氛700 ℃和900 ℃下煅烧1 h 后合成.从图22(a)和(c)中的XRD 图谱可以发现两样品分别为纯P2 和O3 结构,SEM 插图也展现出不同的形貌,其中P2 相具有明显的六边形片状形貌跟传统的P2 相形貌相一致.结果证明同一个组成可以存在两个或两个以上的稳定结构,即有多种晶型存在(例如C 单质,可以以石墨、金刚石、C60等晶型存在.)因此,在层状氧化物材料的合成过程中,仍需对原料、煅烧温度和煅烧时间进行适当控制.电化学测试表明P2 相首周充电比容量114.7 mAh/g,可逆比容量151.09 mAh/g;O3相首周充电比容量134.01 mAh/g,可逆比容量157.47 mAh/g.由于两种材料前几周的可逆比容量和循环相差不大,研究人员认为P2 和O3 相结构对材料的性能影响较少,但是长循环性能和倍率性能文中并没有仔细研究对比. 图22 (a)P2-和(c)O3-Na2/3Fe2/3Mn1/3O2 样品的XRD 谱和SEM 图片;(b) P2-和(d)O3-Na2/3Fe2/3Mn1/3O2 样品前两周在1.5—4.2 V 电压范围内的充放电曲线对比[71]Fig.22.XRD patterns and SEM images of (a) P2-and (c) O3-Na2/3Fe2/3Mn1/3O2 samples;comparison of charge-discharge capacity of (b) P2-and (d) O3-Na2/3Fe2/3Mn1/3O2 within the 1 st and 2 nd cycles in the voltage range of 1.5—4.2 V[71]. 合成时降温速度对材料结构也有一定程度的影响.在锰基层状氧化物中,这一影响更加明显.Parant 等[72]提出在降温过程中Na0.67MnO2会继续与O2反应,锰离子氧化并在结构中形成锰空位,具体反应化学式为Na0.67MnO2+y/2 O2→ Na0.67MnO2+y(Na0.67/(1+y/2)Mn1/(1+y/2)□(y/2)/(1+y/2)O2,□为锰空位).Paulsen 等[73]通过热重测试证明该材料的质量会随着升温和降温过程可逆地减小和增大,这主要跟氧气的释放和吸收有关.研究结果表明锰空位和价态调整都是在缓慢降温过程中发生的.Komaba 课题组[36]通过对P2-Na2/3MnO2材料高温淬火制备了纯相的正交晶系的P′2-Na2/3MnO2材料,材料中锰离子平均价态降低,没有锰空位.正因为结构中存在更多具有姜泰勒效应的Mn3+,晶体结构发生扭曲并从六方晶系转变为正交晶系.后续电化学性能和原位表征测试发现P′2-Na2/3MnO2材料拥有更加可逆的相转变和提高的电化学性能.最近,杨勇课题组[74]提出了通过液氮处理制备无Mn 空位的Mn 基层状氧化物正极材料,如图23 所示.该方法表明液氮淬火能够很好地稳定Mn 基层状氧化物材料的结构并提高其比容量.此外,进一步通过引入廉价的Al 和Fe 离子共掺杂调整Mn 离子的价态,并抑制Na+脱出/嵌入过程中的结构转变,最终获得了良好的循环性能. 图23 不同冷却方式对材料晶体结构中空位的影响示意图[74]Fig.23.Schematic illustration of the effects of different cooling methods on the vacancies of the crystal structures[74]. 笔者课题组分别通过淬火和自然冷却的方法合成相同初始组分的O3-Na0.9Cu0.11Ni0.11Fe0.30Mn0.48Ti0.10O2正极材料[75],经实验发现淬火得到的材料具有更高的体相钠含量(约0.9)和较少的表面碳酸钠.通过原位变温XRD 表征分析了该设计组成在缓慢降温过程中的实时结构演化,发现缓慢降温时材料体相的钠离子会和环境中的二氧化碳以及氧气反应,在样品表面形成无定形的Na2CO3,同步地晶格内的Mn3+发生氧化.相反地,淬火处理不仅能稳定晶格氧框架的长程有序,而且能够降低体相钠离子的脱出和Mn3+的氧化.因此,淬火样品因具有额外的Mn3+电化学氧化,表现出较高的首周充电比容量.再通过控制放电截止电压(2.5 V)避免Mn4+的还原,可以利用该非对称的氧化还原电对实现对全电池首周充电时不可逆钠离子损失的补偿.同时由于抑制了表面Na2CO3的形成,该材料在半电池和全电池中表现出优异的电化学性能.该策略为设计功能型钠离子电池正极材料提供新的思路,为充分发挥过渡金属离子的作用找到一种新的方案. O3 和P2 相氧化物材料是目前报道的最广泛的两类钠离子层状正极材料.O3 相材料一般存在空气稳定性差和Na+扩散动力学较差(Na+在钠层的八面体位之间移动时需要经过中间的四面体空位)的劣势,但是初始相中钠含量高,首周充电可逆比容量高,与不含钠的负极(如硬碳[76,77])匹配全电池时具有较高的适配性.P2 相初始材料中钠离子占据三棱柱位置,扩散动力学条件比O3 相好,但是初始相中钠含量较低,当和不含钠的负极匹配时需要额外的钠源补充.因此研究高钠离子含量的P2 相正极材料具有十分重要的意义.在P2 相材料中,高的钠含量不仅能增强框架结构的稳定性,而且能促进活性离子在较低电压下发生氧化还原反应提供电荷补偿.笔者课题组设计了组成为P2-Na45/54Li4/54Ni16/54Mn34/54O2的化合物[78].结果表明,该组成为纯P2 相结构,高的起始钠含量能够影响结构中O(2p)和TM(3d-eg*)轨道之间的杂化状态,促使材料在2.0—4.0 V 的电压范围内完成Ni2+到Ni4+的两电子氧化还原反应,提供100 mAh/g 的可逆比容量.同时,高的起始钠含量能够调节结构中过渡金属离子的局域环境以及NaO2和TMO2层之间的相互作用,缓解结构由P2 相向O2 相的转变,从而实现较长的循环性能(3000 周).对比于以往的低钠含量的P2 相材料,高钠P2 相材料为研发新的电极材料提供了材料电子结构和化学结构上的新见解. 近日,Sun 等[79]提出构建体相O3-NaNi0.5Mn0.5O2和表面P2-Na2/3MnO2的异质结构正极材料,希望能够兼顾O3 相的高容量和P2 相优异的动力学特性.研究者通过湿化学合成法成功将P2-Na2/3MnO2包覆到O3-NaNi0.5Mn0.5O2正极材料表面.其中,O3 相核心提供了足够的初始Na+储量,而P2 型结构充当了坚固的保护层,均匀的保护层紧密地锚定在主体O3 材料的表面,通过平滑相结构演化和降低晶格应力来增强所得正极的结构完整性和稳定性.同时,电化学动力学分析表明P2-层提供快速Na+扩散通道,促进电荷转移并提高倍率能力.因此,异质结构的正极材料表现出显著提高的电化学性能.然而目前O3 和P2 相两种结构在高电压下的相转变仍决定着结构的稳定性,为了使材料长循环性能满足实际需要仍需要从本质上解决相转变不稳定的问题. 经过近些年来的发展,钠离子电池综合性能和示范应用均取得了显著的成效.钠离子电池作为锂离子电池的有益补充将在大规模储能和微型电动车领域取得长足发展.高比能量的电极材料仍将是人们孜孜以求的目标,然而钠离子层状氧化物正极材料在实现更多钠离子可逆脱出/嵌入时,体系面临复杂的结构演变,严重影响其长循环过程中的容量保持率.例如:1)复杂的相转变,P2 或O3 正极材料在体相钠离子含量不足1/3 时,将发生P 型结构向O 型结构的演变并引起较大的体积变化,进一步会诱导晶间裂纹的产生和过渡金属离子溶解;2)不可逆的过渡金属离子迁移,尤其是富含Fe 和Cr 的材料,在体系中的钠离子含量较少时晶格能发生显著变化,致使过渡金属离子迁移到钠层的四面体位或者八面体位;3)电解液副产物的共嵌入,高度缺钠态的O 型结构化学稳定性显著降低,会引发电解液分解的小分子产物进入层状晶格,导致活性储钠位点的损失. 尽管对过渡金属位掺杂或替代可以显著抑制相转变、抑制过渡金属离子的迁移和提高脱钠态材料的化学和电化学稳定性,但会不可避免地降低体系氧化还原电对的含量进而影响体系能量密度,且掺杂离子含量太少将不足以稳定层状氧化物材料的结构.另外,可能通过对钠离子位进行掺杂K+,Mg2+,Ca2+或Zn2+,起到支柱作用,从而有效缓解钠层O-O 之间的静电排斥作用,抑制相转变的发生,但是掺杂量不易过大,以免牺牲体系的能量密度.包覆改性在锂离子层状氧化物正极材料的应用较多且效果显著,但是在钠电层状氧化物中的研究还较少.主要是因为钠离子层状氧化物的空气和水稳定性较差,这使得在锂电中常用的水溶液包覆方法在钠电中不再适用,而且复杂的操作流程会显著提高电极材料的制备成本.因此发展简单有效且可以提高钠电层状氧化物材料在较高比容量下的循环性能的包覆方法势在必行. 本文详细介绍了钠离子层状氧化物材料的结构及其演变规律,尤其是高电压下的结构演化过程.通过深入理解组成和结构演变之间的关系以及对电化学性能的影响,为合理设计下一代正极材料提供新的见解.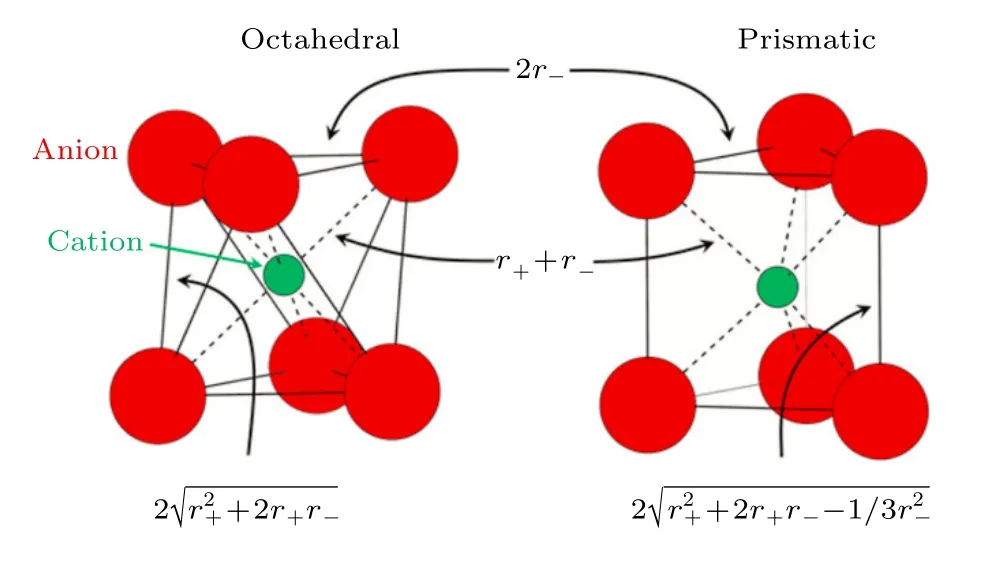
3.1 O3 相转变





3.2 P2 相转变








4 钠离子层状氧化物常见组成的结构性能分析
4.1 元素组成对相变以及电化学性能的影响



4.2 合成条件对材料结构及电化学性能的影响

4.3 初始钠含量对电化学性能的影响
5 总结与展望
猜你喜欢
仪器仪表用户(2021年10期)2021-11-27 08:26:14陶瓷学报(2020年6期)2021-01-26 00:37:56重型机械(2019年3期)2019-08-27 00:58:44中学生数理化·中考版(2018年11期)2019-01-31 06:18:06教学考试(高考化学)(2018年5期)2018-12-06 07:21:56焊接(2016年9期)2016-02-27 13:05:22中华老年多器官疾病杂志(2016年2期)2016-01-16 03:15:46新疆钢铁(2015年2期)2015-11-07 03:27:52电源技术(2015年2期)2015-08-22 11:28:30物理化学学报(2015年5期)2015-02-28 17:34:57
