
蛋白C p.Ala333Thr突变引起遗传性蛋白C缺陷症的分子机制
2022-05-31 05:47余晓敏常国林郑逸旸陈诚唐施艺吴鑫媛吕佳林向阳朱丽青
温州医科大学学报 2022年5期
余晓敏,常国林,郑逸旸,陈诚,唐施艺,吴鑫媛,吕佳,林向阳,朱丽青
1.温州医科大学附属第一医院 医学检验中心 浙江省检验诊断及转化研究重点实验室,浙江 温州 325015;2.温州医科大学附属第一医院 温州市血液学重点实验室,浙江 温州 325015;3.温州医科大学附属第二医院 病理科,浙江 温州 325027
遗传性蛋白C缺陷症(inherited protein C deficiency, IPCD)是由位于2号染色体2q13-2q14的蛋白C基因(protein C gene, PROC)突变所引起的常染色体隐性、显性或不完全显性遗传性疾病[1],全球发病率约为1/16 000[2]。IPCD的典型临床症状包括血栓性并发症、暴发性紫癜、弥漫性血管内凝血和新生儿期继发性出血,与PROC突变类型密切相 关[3]。截至目前,已报道300多种PROC突变[4]。IPCD可分为两种类型:I型表现为蛋白C(protein C, PC)抗原水平和功能同时降低,II型表现为PC功能降低而抗原水平正常,或PC活性远低于抗原水平,大多数IPCD属于I型缺陷[5]。虽然IPCD于1981年就曾被首次报道[6],但其具体致病机制至今尚未完全明晰。本课题组发现了1个由p.Ala333Thr突变引起的I型IPCD家系,家系成员中多位有血栓形成史[7],经测序发现患者PROC的第9号外显子存在c.1084G>A杂合错义突变,导致p.Ala333Thr,但基因突变导致PC含量降低的分子机制尚不明确。因此,本研究构建PC p.Ala333Thr突变型质粒,通过体外实验对PC p.Ala333Thr突变导致IPCD的致病机制进行探讨。
1 材料和方法
1.1 先证者及其家系 先证者及其家系6人均纳入研究,随机抽取本院100名健康成年人作为对照组,其中男45例,女55例,年龄12~58岁。本研究通过温州医科大学附属第一医院的伦理委员会审查(伦理批号:KY2022-R050),且所有受试者均知情并签署知情同意书。先证者,女,39岁,因双下肢反复疼痛前往温州医科大学附属第一医院就诊。既往有下肢深静脉血栓形成史,长期口服华法林抗凝,家族中父亲因脑静脉窦血栓死亡,哥哥因肺栓塞死亡,家系图见图1。入院检查:PC活性39%,抗核抗体1∶320阳性,下肢血管超声显示左小腿深静脉血栓形成,临床诊断为“PC缺陷症,系统性红斑狼疮,下肢深静脉血栓形成”。

图1 IPCD家系图
1.2 人胚胎肾细胞HEK-293T的培养 HEK-293T细胞培养于含有10%胎牛血清(美国Gibco公司)的DMEM完全培养基。当细胞生长密度达到80%~90%时以1∶5的比例进行传代,传代后将细胞置于37 ℃、 5% CO2培养箱静置培养,接种后2~3 d即可长成致密单层。
1.3 PC p.Ala333Thr突变体质粒的构建 在已有野生型质粒:pCMV3-SP-N-Flag-PC WT(北京Sino Biological公司,见图2)的基础上,采用 QuickMutationTM定点突变试剂盒(上海碧云天生物技术有限公司)构建突变体表达质粒:pCMV3-SP-NFlag-PC p.Ala333Thr。挑取多个克隆进行全长测 序,经测序发现定点突变成功引入,且没有引入其他变异,最后将构建成功的突变质粒用QIAGEN质粒提取试剂盒(北京天根生化科技有限公司)大规模抽提,并转染入生长状态佳的HEK-293T细胞进行过表达。所有质粒都经正、反向测序证实构建成功,质粒测序图谱见图3。

图2 质粒结构图

图3 PROC第9号外显子存在c.1084G>A杂合错义突变
1.3 细胞转染 转染实验分为WT组(转染pCMV3-SP-N-Flag-PC WT质粒)和Ala333Thr组(转染pCMV3-SP-N-Flag-PC p.Ala333Thr质粒),每组3个复孔,按Lipofectamine 2000试剂(美国ThermoFisher公司)说明书进行瞬时转染。将生长状态佳的HEK-293T细胞按5×105/孔接种于6孔培养板中,每孔加入3 mL无双抗完全培养基(含10%胎牛血清),在37 ℃,5% CO2的恒温培养箱中培养16 h至细胞密度达60%~70%时进行转染。分别在转染24 h后提取细胞总RNA进行RT-qPCR分析,转染48 h后提取细胞总蛋白质进行Western blot分析并收集细胞培养上清液进行ELISA分析。
1.4 RT-qPCR检测PC转录水平 使用RNA提取试剂盒(北京天根生化科技有限公司)抽提转染细胞总RNA,用PrimeScript RT Reagent Kit(日本TaKaRa 公司)配制RT反应液,混匀后进行反转录反应,使用ABI Prism 7500实时荧光定量PCR仪(美国 Applied Biosystems公司),按照两步法PCR扩增程序执行,反应结束后确认RT-qPCR的扩增曲线和熔解曲线。采用Ct法比较分析突变型PROC和野生型PROC表达水平的差异。
1.5 Western blot检测细胞内PC表达水平 提取收集细胞总蛋白后,通过Western blot法检测细胞系中PC表达量。SDS-PAGE进行蛋白分离,恒压电流浓缩胶80 V,分离胶120 V。湿转法进行蛋白转膜,冰浴中以300 mA电流转膜90 min。用5%的脱脂奶粉室温封闭1 h。按照说明书标示比例加入经稀释的PC抗体(美国Proteintech公司)、GADPH抗体(美国Proteintech公司),4 ℃轻摇过夜。次日,洗膜后按照1∶5 000体积比加入PBST稀释的兔抗,室温轻摇1 h。最后将曝光液滴加在条带上,置于曝光机内曝光,使用软件Image J分析获得的条带。
1.6 ELISA检测细胞外PC表达水平 转染48 h后,分别收集两组所有细胞培养液,用Amicon Ultra-4 10K超滤管将细胞培养液浓缩10倍。通过ELISA法检测细胞外PC表达水平。按照说明书(上海江莱生物科技有限公司)稀释标准品至相应浓度,在标准品孔内加入不同浓度的标准品50 μL,样本孔中加入待测样本50 μL,空白孔中加样本稀释液50 μL。除空白孔外加辣根过氧化物酶标记抗体100 μL/孔,封板后置于37 ℃温育60 min。洗涤液重复洗板5次后加入底物A,B各50 μL,37 ℃避光孵育15 min后加入终止液50 μL,15 min内在450 nm波长处测定OD值。计算所得的样本浓度,除以浓缩倍数即WT组和Ala333Thr组PC的细胞外浓度。
1.7 PC及细胞器免疫荧光染色 铺Ф35 mm共聚焦专用皿,每皿接种5×104个细胞。细胞置于37 ℃、5% CO2培养箱培养24 h后转染,继续培养48 h。一抗使用3% BSA配制PC兔多抗(1∶500),二抗使用3% BSA配制抗兔FITC抗体(1∶200)。按照说明书分别配制ER Tracker Red工作液(上海碧云天生物技术有限公司)(1∶2 000)和Golgi Tracker Red工作液(上海碧云天生物技术有限公司)(1∶800),室温下避光孵育1 h吸走所有工作液,使用PBS清洗3次。在荧光显微镜下观察PC在内质网(endoplasmic reticulum, ER)和高尔基体(Golgi apparatus, GA)中的分布情况。
1.8 统计学处理方法 采用SPSS23.0软件进行统计学分析。计量资料采用±s表示,两组均数比较采用独立样本t检验。P<0.05为差异有统计学意 义。
2 结果
2.1 生物信息学分析 PolyPhen-2是用于预测氨基酸替代对蛋白质结构和功能影响的软件,通过给出预测分值量化评估突变类型的有害性,该值越接近1则表示突变造成的危险性越高,PolyPhen-2对先证者PC的突变类型危害程度评分为0.763,因而,预测p.Ala333Thr有害性为高。此外,我们利用多序列在线比对工具T-Coffee对PC的第333位氨基酸在同源物种间的保守性进行评估,显示该位点保守性不高,见图4。

图4 PC第333位氨基酸保守性分析结果
2.2 PC p.Ala333Thr突变体转录水平和细胞内外蛋白水平变化 RT-qPCR结果显示,转染24 h后,PC WT和PC Ala333ThrPROCmRNA表达差异无统计学意义(P=0.120),表明PC Ala333Thr突变体在转录水平未发生明显变化(见图5A)。Western blot结果显示,质粒转染48 h后PC WT和PC Ala333Thr的PC蛋白表达差异有统计学意义(P=0.020),表明PC Ala333Thr突变体在HEK-293T细胞中的蛋白表达量明显降低(见图5B)。转染48 h后,收集培养细胞的上清液,通过ELISA检测上清液中PC含量,实验结果显示p.Ala333Thr突变体的PC水平为60%±27.4%,明显低于WT组,这与I型IPCD的临床表型一致(见图5C)。
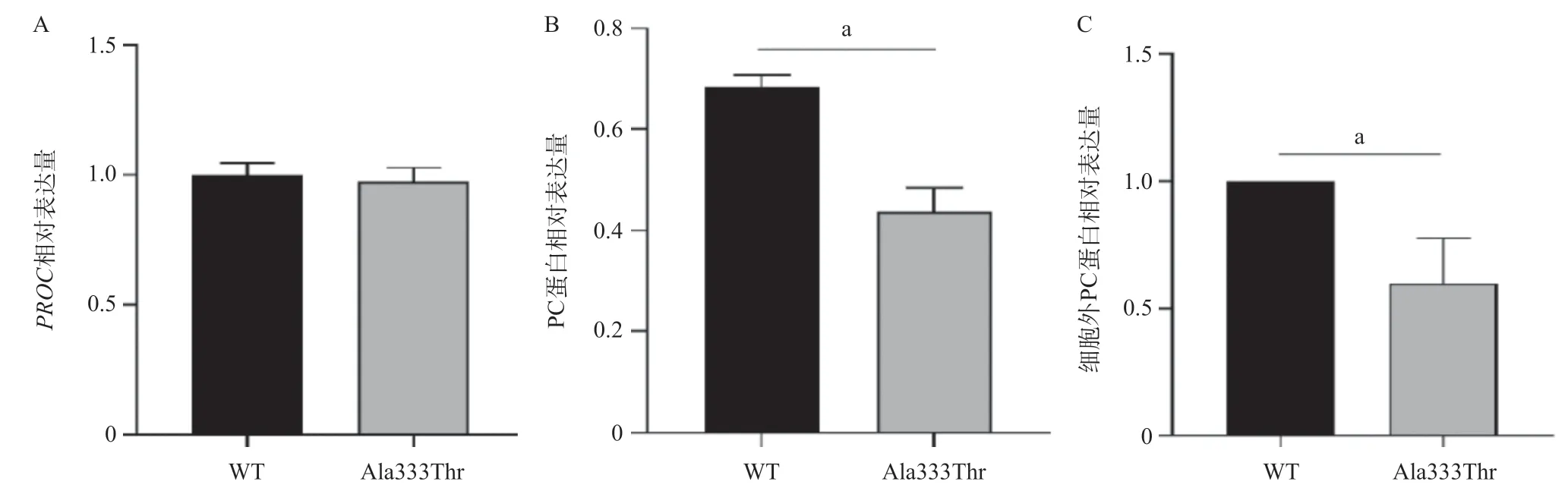
图5 PC p.Ala333Thr突变体转录水平和细胞内外蛋白水平变化
2.3 PC p.Ala333Thr突变体在ER和GA中含量的变化 我们将野生型PC质粒和p.Ala333Thr突变PC质粒转染后的HEK-293T细胞分别用ER和GA的荧光染料进行染色。染料可以使相应的细胞器在激光共聚焦显微镜下经594 nm激光激发红色荧光,而经染色的PC经488 nm激光激发绿色荧光,若PC在相应细胞器内,则两种颜色重叠显黄色荧光。结果显示,野生型PC除在ER分布外,在GA内也大量存在;突变蛋白主要分布在ER,少量进入GA内,见图6。
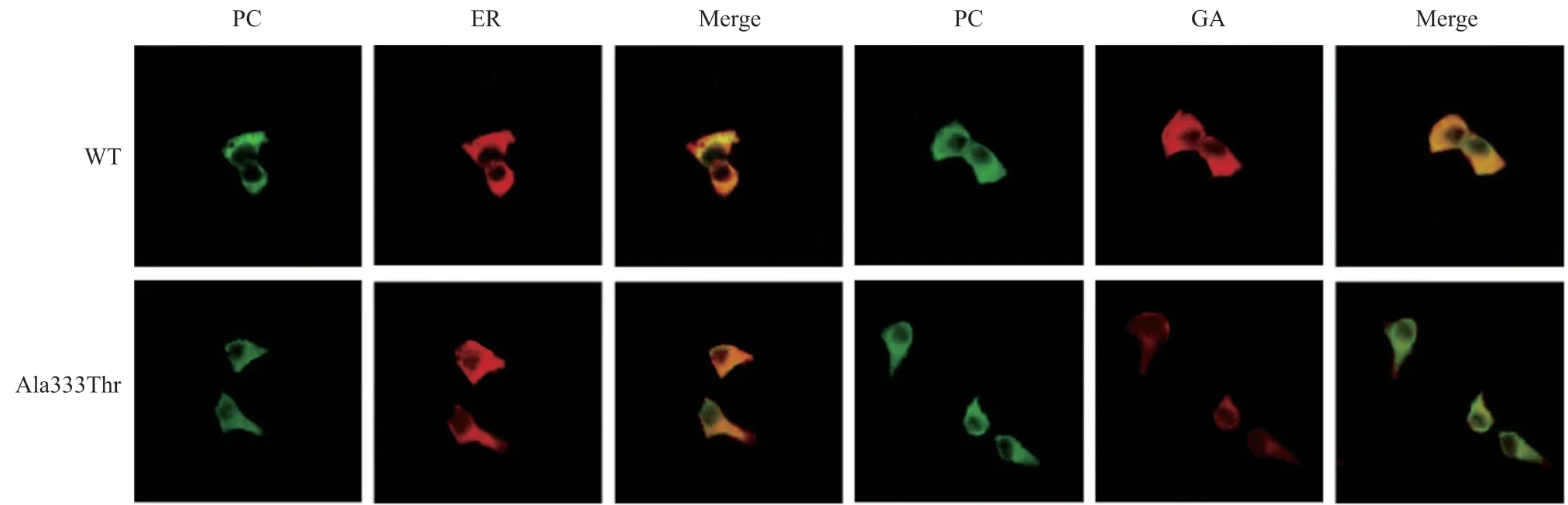
图6 野生型PC和突变型PC在细胞器内的定位
3 讨论
PC是人体重要的抗凝蛋白,在机体抗凝血功能中发挥重要作用。PC在凝血酶或凝血酶-血栓调节蛋白复合物的催化下,转变为活化蛋白C(activated protein C, APC),后者与辅因子蛋白S(protein S, PS)结合后,通过多种机制发挥抗凝血作用[8]。PC缺乏可引起静脉血栓栓塞、暴发性紫癜、弥散性血管内凝血等。
IPCD是由2号染色体长臂(2q13-14)上的PC基因突变引起[9],多为常染色体显性遗传,严重的PC缺陷与纯合性或复合杂合性突变有关,最终通过影响mRNA的正常序列和蛋白质合成导致IPCD[10]。在我们的研究中,先证者及其家系发生PC p.Ala333Thr 突变,导致IPCD发生,引起机体静脉血栓栓塞等多种疾病。
本研究成功构建了PC p.Ala333Thr突变的质粒,经细胞转染后发现该突变导致PC表达量相对下降,该结论与临床结果相符合[7]。因此认为患者PROC的p.Ala333Thr突变导致PC含量偏低。为探讨p.Ala333Thr突变位点对PC结构和功能的影响,我们通过PolyPhen-2软件对p.Ala333Thr突变进行评估,评分结果为0.763分,评估有害性高,提示该突变可能影响到PC的结构和功能。为进一步分析p.Ala333Thr突变引起的IPCD的分子致病机制,本研究用野生型质粒和p.Ala333Thr突变型质粒分别转染HEK293T细胞。RT-qPCR实验显示,p.Ala333Thr突变型质粒转染的细胞PC mRNA转录水平与野生型质粒转染的细胞PC mRNA相比没有明显差别,说明该位点的突变并没有导致PROC基因在转录水平的下降,而Western blot和ELISA结果显示,细胞内和细胞外的PC都出现显著降低,因此,蛋白表达量的下降原因可能发生在细胞内细胞器对蛋白质进行加工、转运等过程中。我们选择了在细胞内负责蛋白质加工、转运和筛选的两种主要细胞器ER和GA,进行免疫荧光染色。免疫荧光染色结果显示,PC p.Ala333Thr突变体在ER中的含量变化不明显,而在GA内突变体含量相对野生型明显下降。因此我们推测PROC的p.Ala333Thr突变可能导致其翻译或PC转运过程受损。
ER是一种连接细胞核、细胞质和细胞膜的细胞器,在生物合成、折叠、稳定、分泌和跨膜蛋白运输中发挥重要作用。值得重视的是,蛋白质的折叠和组装过程很容易发生错误,导致错误折叠蛋白质的产生,引起蛋白质稳态失衡[11]。为了维持稳定,ER通过泛素-蛋白酶体系统或溶酶体来筛选和清除部分错误折叠的蛋白质,称之为ER相关降解或ER吞 噬[12-13]。在本研究中,PROC发生了p.Ala333Thr位点的突变,该突变可能导致PC的折叠与组装发生错误,从而引发PC在ER内被降解,不能顺利运输入GA内。
GA是细胞内中心的膜结合细胞器,在分泌蛋白及脂类的运输、加工和分类中起关键作用[14]。错误折叠的蛋白质可脱离ER而被运输到GA,由GA对其进行识别,由溶酶体靶向降解[15]。在本研究中,免疫荧光结果显示,PC p.Ala333Thr突变体在GA内的含量明显下降,证实了PC突变体含量的下降是在ER向GA运输的过程中发生的。
综上所述,本研究证实PC p.Ala333Thr突变造成PC表达量的下降,从而引发IPCD。通过体外表达实验,我们推测:PC表达量下降的关键分子机制在于p.Ala333Thr位点突变造成PC发生了折叠错误,从而在ER内被分选并促使错误折叠的PC被降解或吞噬,阻止其向GA运输,最终导致PC含量的减少。
猜你喜欢
作物学报(2022年2期)2022-11-06
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
作物学报(2022年8期)2022-05-29
蔬菜(2021年7期)2021-11-27
科学24小时(2021年4期)2021-03-22
科学导报(2020年70期)2020-11-09
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
