
菊花含量测定方法提高
2022-05-30 21:00王小芳郑倩倩路丽娟
中国民族民间医药·上半月 2022年1期
王小芳 郑倩倩 路丽娟
【摘 要】 目的:改进《中国药典》中菊花含量测定方法,使其中3,5-O-二咖啡酰基奎宁酸与其同分异构体1,5-O-二咖啡酰基奎宁酸得到良好分离,提高测定结果准确性。方法:Inertsil ODS-3 C18色谱柱(4.6 mm×250 mm,5 μm),流动相为乙腈-甲醇-0.2%冰醋酸溶液梯度洗脱,检测波长为348 nm,柱温30 ℃,流速1 mL/min,进样量10 μL。结果:绿原酸、木犀草苷和3,5-O-二咖啡酰基奎宁酸分离良好,在12.583~314.582、12.595~314.878、15.599~389.978 μg/mL的范围内呈现良好的线性关系(r=0.9999、1.0000、0.9995);平均加样回收率分别为98.21%、102.18%、97.77%,RSD为1.54%、0.99%、1.01%(n=6)。结论:该方法专属性强,准确、灵敏,解决了现行方法的问题。
【关键词】 菊花;含量测定;3,5-O-二咖啡酰基奎宁酸
【中图分类号】R927.2 【文献标志码】 A 【文章编号】1007-8517(2022)01-0044-05
Improve Chinese Pharmacopoeia Contentdetermination of Chrysanthemum Morifolium Ramat.
WANG Xiaofang ZHENG Qianqian LU Lijuan
Shijiazhuang Institute for Food and Drug Control,Shijiazhuang 050011, China
Abstract:Objective To improve contentdetermination of Chrysanthemum morifolium Ramat. in the current Chinese Pharmacopoeia to well separate 3,5-O- Dicaffeoyl quinic acid. Methods Inertsil ODS-3 C18 (4.6×250 mm,5 μm) ,with the mobil phase comprising of acetonitrile- methanol -0.2%ice acetate flowing at 1mL/min in a gradient elution manner, and the detection wavelengths were set at 348 nm,and the column temperature was 30 ℃.Results The linear ranges of chlorogenic acid, luteolin, 3,5-O-dicaffeoyl quininic acid fell into 12.583~314.582 μg/mL (r=0.9999)、12.595~314.878 μg/mL(r=1.0000)、15.599~389.978 μg/mL(r=0.9995)respectively.The average recovery of chlorogenic acid, luteolin, 3, 5-O-dicaffeoyl quininic acid were 98.21%、102.18%、97.77%with the RSDs of 1.54%、0.99%、1.01%(n=6).Conclusion The new mothed is specific,accurate and sensitive. It solves the problems of the current Chinese pharmacopoeia mothed.
Key words:
Chrysanthemum Morifolium Ramat.; Content Eetermination; 3,5- Dicaffeoylquinic Acid
菊花為菊科植物菊Chrysanthemum morifolium Ramat.的干燥头状花序。具有散风清热、平肝明目、清热解毒的功效,用于风热感冒、头痛眩晕、目赤肿痛、眼目昏花、疮痈肿毒。药理研究表明菊花具有抗病毒、抑菌、抗炎、抗氧化等药理作用,其主要成分为黄酮类、有机酸类、挥发油类等。
该品种现行标准为2020版《中国药典》,含量测定以绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸3个成分作为其质控成分,采用高效液相色谱法,以乙腈-0.1%磷酸溶液进行梯度洗脱。但在日常检验中发现,采用药典方法测定含量时3,5-O-二咖啡酰基奎宁酸与其同分异构体1,5-O-二咖啡酰基奎宁酸不能有效分离,导致计算结果偏差较大,不能真实反应该成分含量,需要对现有方法进行改进。虽然近年来关于菊花多成分测定的报道较多,但并未检索到能将二者有效分离的高效液相色谱方法。因此本实验对菊花含量测定方法进行改进,通过改变流动相对色谱条件进行改进和优化,实现对3,5-O-二咖啡酰基奎宁酸的有效分离,从而提高检测结果的准确性。
1 仪器与试药
1.1 仪器 Waters ARC高效液相色谱仪(美国Waters公司);CPA225D电子天平(德国赛多利斯公司);Elmasonic P 超声波清洗仪(德国Elma公司);水浴锅(天津泰斯特)。
1.2 试药 绿原酸对照品(批号:110753-201716,纯度以99.3%计)、木犀草苷对照品(批号:111720-201408,纯度以94.8%计)、3,5-O-二咖啡酰基奎宁酸对照品(批号:111782-201706,纯度以97.3%计)均购自中国食品药品检定研究院;1,5-O-二咖啡酰基奎宁酸(批号:E-0447-18042523,上海同田生物技术股份有限公司)。乙腈、甲醇、冰醋酸、磷酸均为色谱纯;水为超纯水。4批菊花(购自市场)经本中心毕飞霞老师鉴定为菊科植物菊 Chrysanthemum morifolium Ramat.的干燥头状花序,样品1为杭白菊、样品2~4为贡菊。
2 方法与结果
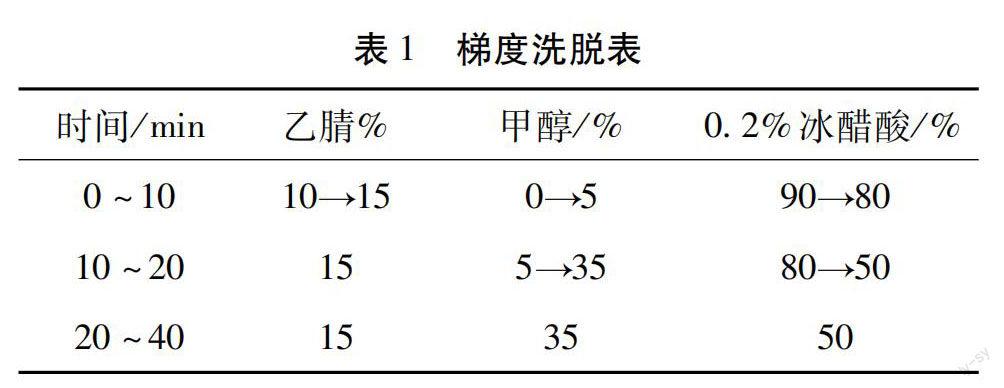
2.1 色谱条件及系统适用性 试验Inertsil ODS-3 C18色谱柱(4.6 mm×250 mm,5 μm),流动相为乙腈-甲醇-0.2%冰醋酸溶液梯度洗脱,程序见表1,检测波长为348 nm,柱温30 ℃,流速1 mL/min,进样量10 μL。
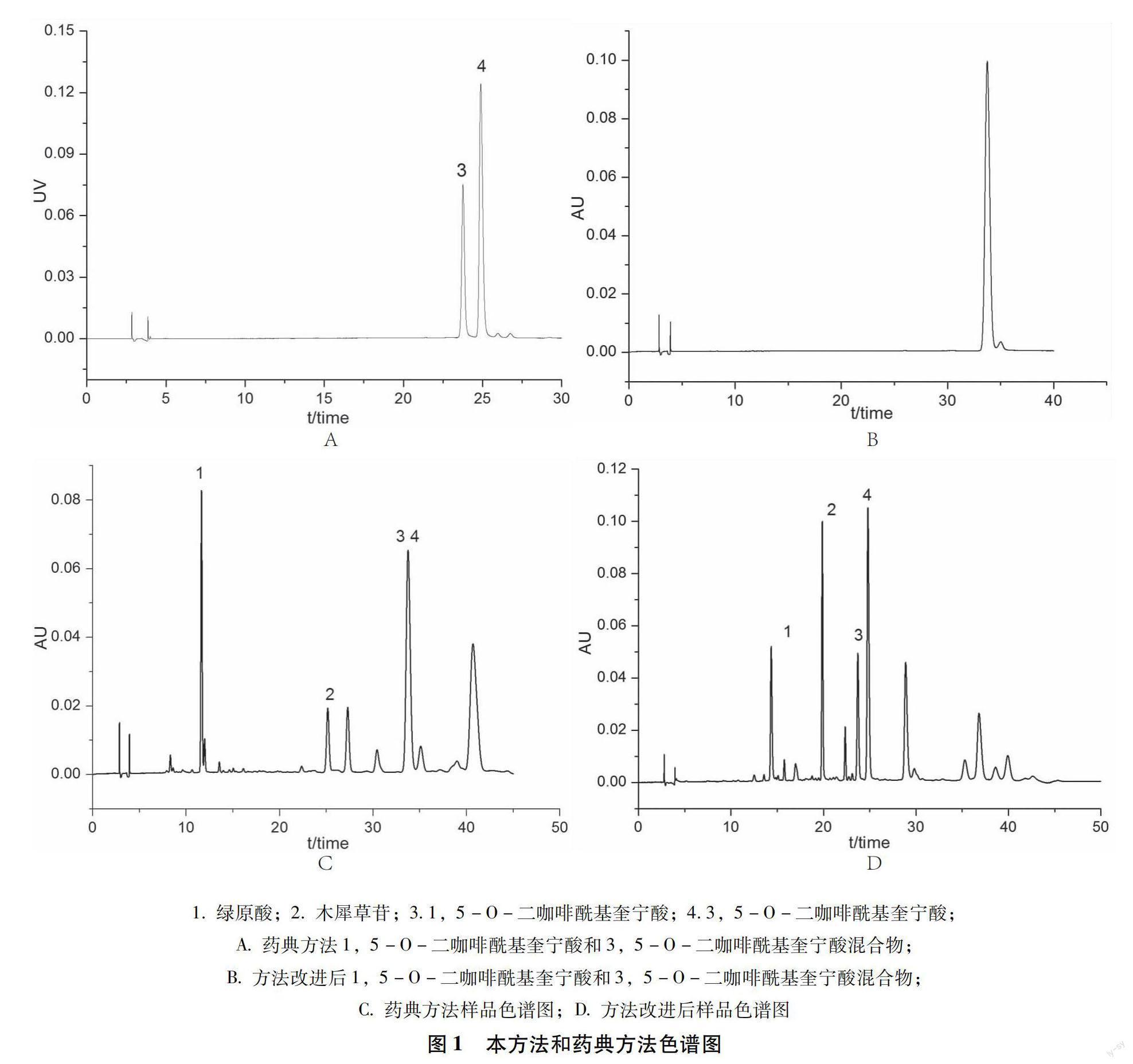
本方法,绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸各峰的理论板数均大于10000,分离度均大于1.5。本方法和药典方法条件下,对照品和供试品色谱图如图1所示。
2.2 溶液制备
2.2.1 对照品储备液和混合对照品溶液的制备 分别精密称取绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸对照品适量,加70%甲醇分别制成含绿原酸1.5729 mg/mL、木犀草苷1.5744 mg/mL、3,5-O-二咖啡酰基奎宁酸1.9499 mg/mL的对照品储备液。分别精密吸取上述储备液各1 mL,用70%甲醇定容至25 mL,即得混合对照品溶液。
2.2.2 供试品溶液的制备 取菊花粉末(过一号筛)约0.25 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25 mL,密塞,称定重量,超声处理(功率300 W,频率45 kHz)40 min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3 方法学评价
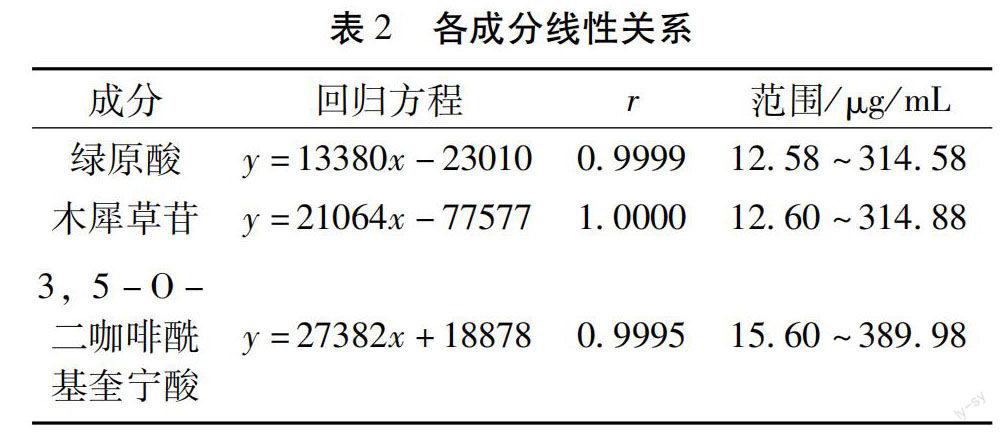
2.3.1 线性关系考察 分别精密吸取2.2.1项下的各对照品储备液1 mL置5、10、25、50 mL量瓶、2 mL置250 mL量瓶,加70%甲醇定容至刻度,摇匀,即得稀释5、10、25、50、125倍的5 个浓度梯度的线性考察混合对照品溶液,按照2.1项下色谱条件进行测定,记录峰面积。以对照品浓度为横坐标(X),以峰面积为纵坐标(Y),绘制标准曲线,结果见表2,可知各成分在各自范围内线性关系良好。
2.3.2 精密度考察 取2.3.3项下制备的样品溶液1份,按照2.1项下色谱条件进行测定,连续进样6次,记录色谱图峰面积,测得绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸峰面积的RSD值分别为0.85%、0.44%、0.52%,表明仪器精密度良好。
2.3.3 重复性考察 取同一批供试品,按“2.2.2”项下方法平行制备6份供试品溶液,在“2.1”色谱条件下进样测定,测得6份样品中绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸含量的RSD值分别为0.64%、1.41%、1.03%,表明該方法重复性良好。
2.3.4 回收率试验 精密称取同一批供试品约0.125 g,共6份,置于具塞锥形瓶中,精密加入各对照品,按“2.2.2”项下方法平行制备6份供试品溶液,在“2.1”色谱条件下进样测定,计算回收率。结果见表3,结果说明该方法的准确度良好。
2.3.5 稳定性考察 取2.3.3项下制备的样品溶液1份,按照2.1项下色谱条件进行测定,分别在0、2、6、8、16、24 h进样,记录色谱图,计算3种成分峰面积的RSD值,绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸含量的RSD值分别为1.25%、0.66%、0.55%,表明该方法稳定性良好。
2.3.6 色谱柱耐用性试验 取同一批样品,照“2.3.3”项下方法制备供试品溶液,对不同品牌色谱柱的耐用性进行考察,并计算含量。其中Agilent 5Tc-C18(2)(4.6 mm×250 mm,5 μm)色谱柱在2.1色谱条件下木犀草苷和3,5-O-二咖啡酰基奎宁酸的分离度均小于1.5。TechMate C18-ST(4.6 mm×250 mm,5 μm)、Inertsil ODS-3(4.6 mm×250 mm,5 μm)系统适用性较好,结果见表4。
2.3.7 样品的测定 按2.2.2项下方法,制备4批菊花供试品溶液,按2.1项下色谱条件和药典方法分别进行含量测定,记录峰面积,按干燥品计算样品中绿原酸、木犀草苷和3,5-O-二咖啡酰基奎宁酸的含量。结果见表5。
3 讨论
3.1 分析条件的优化 实验过程中首先选用了3种不同品牌色谱柱Agilent 5Tc-C18(2)(4.6 mm×250 mm,5 μm)、TechMate C18-ST(4.6 mm×250 mm,5 μm)、Inertsil ODS-3(4.6 mm×250 mm,5 μm),在药典方法乙腈-0.1%磷酸溶液的基础上调整流动相比例,但3,5-O-二咖啡酰基奎宁酸和1,5-O-二咖啡酰基奎宁酸均重合在一起,无法分离。之后改变流动相组成,采用甲醇-磷酸溶液系统,可使3,5-O-二咖啡酰基奎宁酸和1,5-O-二咖啡酰基奎宁酸分离度大于1.5,但木犀草苷分离度小于1.5。后又试验了乙腈-甲醇-0.1%磷酸系统和乙腈-甲醇-0.2%冰醋酸系统,发现,乙腈-甲醇-0.2%冰醋酸梯度洗脱各成分分离度较好,绿原酸、木犀草苷,3,5-O-二咖啡酰基奎宁酸分离度均大于1.5,最终选择该系统梯度洗脱。
3.2 检测波长的选择 通过扫描200~400 nm波长范围内的紫外吸收,发现绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸的最大吸收分别为326.1、348.8、327.3 nm,绿原酸和3,5-O-二咖啡酰基奎宁酸在327 nm与348 nm处的峰面积比接近1.8,木犀草苷在327 nm与348 nm处的峰面积比接近0.8,如果检测波长选择327 nm对3种成分峰面积影响较小,但是由于木犀草苷的含量较低,峰面积较小,对其准确度影响较大,因此本方法还是采用药典方法的348 nm。
3.3 2020版《中国药典》菊花含量测定 方法中3,5-O-二咖啡酰基奎宁酸与1,5-O-二咖啡酰基奎宁酸不仅出峰时间一致,且二者混合物的紫外吸收与3,5-O-二咖啡酰基奎宁酸也完全一致(如图2所示),在日常检验中很容易误判,造成检验结果的误差。药典菊花中3,5-O-二咖啡酰基奎宁酸的限度为不得少于0.70%,由样品测定结果可以看出,3,5-O-二咖啡酰基奎宁酸的含量前后相差较大,有的甚至会影响其是否合格的判定。相比药典方法,本方法解决了3,5-O-二咖啡酰基奎宁酸的分离问题,检测结果更加可靠。
本实验采用的新方法能较好的分离菊花中的绿原酸、木犀草苷和3,5-O-二咖啡酰基奎宁酸,具有较高的灵敏度和准确度,重现性好,解决了现行方法3,5-O-二咖啡酰基奎宁酸与1,5-O-二咖啡酰基奎宁酸不能分离的问题,可作为该品种含量方法改进的参考。
参考文献
[1]国家药典委员会.中华人民共和国药典(一部)[S]. 北京:中国医药科技出版社,2020:323.
[2]周衡朴,任敏霞,管家齐,等.菊花化学成分、药理作用的研究进展及质量标志物预测分析[J].中草药,2019,50(19):4785-4795.
[3]王德胜,黄艳梅,石岩,等.菊花化学成分及药理作用研究进展[J].安徽农业科学,2018,46(23):9-11.
[4]覃珊,温学森.HPLC同时测定菊花中6种活性成分含量[J].中国中药杂志,2011,36(11):1474.
[5]張维冰,王智聪,张凌怡.超高效液相色谱-二极管阵列检测-串联质谱法测定菊花中的10种咖啡酰基奎宁酸和22种黄酮类化合物[J].分析化学,2013,41(12):1851.
[6]杜憬生,吴立群,刘敬功,等.基于UPLC-Q-TOF-MS技术的菊花化学成分快速分析[J].中药材,2017,40(3):621.
[7]孙婷婷,魏雅平,程红,等.HPLC法同时测定不同栽培区域滁菊中8种成分含量[J].安徽科技学院学报,2019,33(1):34-40.
[8]吕盼,余诗琪,张飞,等.药用菊花中10种化学成分的含量测定及主成分分析[J].中国药师,2018,21(8):1374-1378.
[9]李婷婷,张琴,赖森亚,等.怀菊花多成分含量测定及指纹图谱的模式识别研究[J].中国现代应用药学,2015,32(5):551-556.
[10]姜涛,陈林明,姚艺新,等.3种规格菊花有效成分与重金属元素的质量评价研究[J].中医药导报,2020,26(9):37-44.
[11]李丹霞,王康才,成明超,等.杭白菊6个品种主要化学成分分布规律比较研究[J].中药材,2013,36(8):1231-1234.
(收稿日期:2021-05-06 编辑:陶希睿)
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2020年10期)2020-11-20
分析测试学报(2020年4期)2020-05-09
发酵科技通讯(2018年2期)2018-07-06
天然产物研究与开发(2018年2期)2018-04-04
西江月(2017年4期)2017-11-22
长春中医药大学学报(2017年1期)2017-04-16
小雪花·成长指南(2016年10期)2016-11-01
知音·下半月(2016年5期)2016-05-27
中国野生植物资源(2014年1期)2014-03-29
中成药(2014年10期)2014-02-28
