
cGAS-STING通路与自噬交互作用的研究进展
2022-05-26 02:44阳怡羽张旭飞综述吴秀文任建安审校
医学研究生学报 2022年5期
阳怡羽,张旭飞综述,吴秀文,任建安审校
0 引 言
固有免疫应答中的cGAS-STING通路在抵御病原微生物入侵方面具有重要的作用[1],病原微生物和损伤的宿主细胞可释放游离双链DNA(double strands DNA,dsDNA),胞质DNA传感器环鸟苷单磷酸-腺苷单磷酸合酶(cyclic guanosine monophosphate-adenosine monophosphate synthase, cGAS)识别dsDNA,催化 ATP 和 GTP 合成第二信使cGAMP,从而激活内质网上的干扰素基因刺激因子(stimulator of interferon genes, STING),募集TANK 结合激酶 1(TANK-binding kinase 1,TBK1),导致转录因子干扰素调节因子 3(interferon regulatory factor 3,IRF3)的磷酸化和入核,促进I 型干扰素(interferon,IFN)和炎症因子的表达[2-3],进而增强免疫反应加强宿主防御。同样,自噬也可调节免疫系统的防御功能。自噬可直接捕获并清除病原微生物,也能够与cGAS-STING通路等模式识别受体信号通路相互作用[4]。见图1。
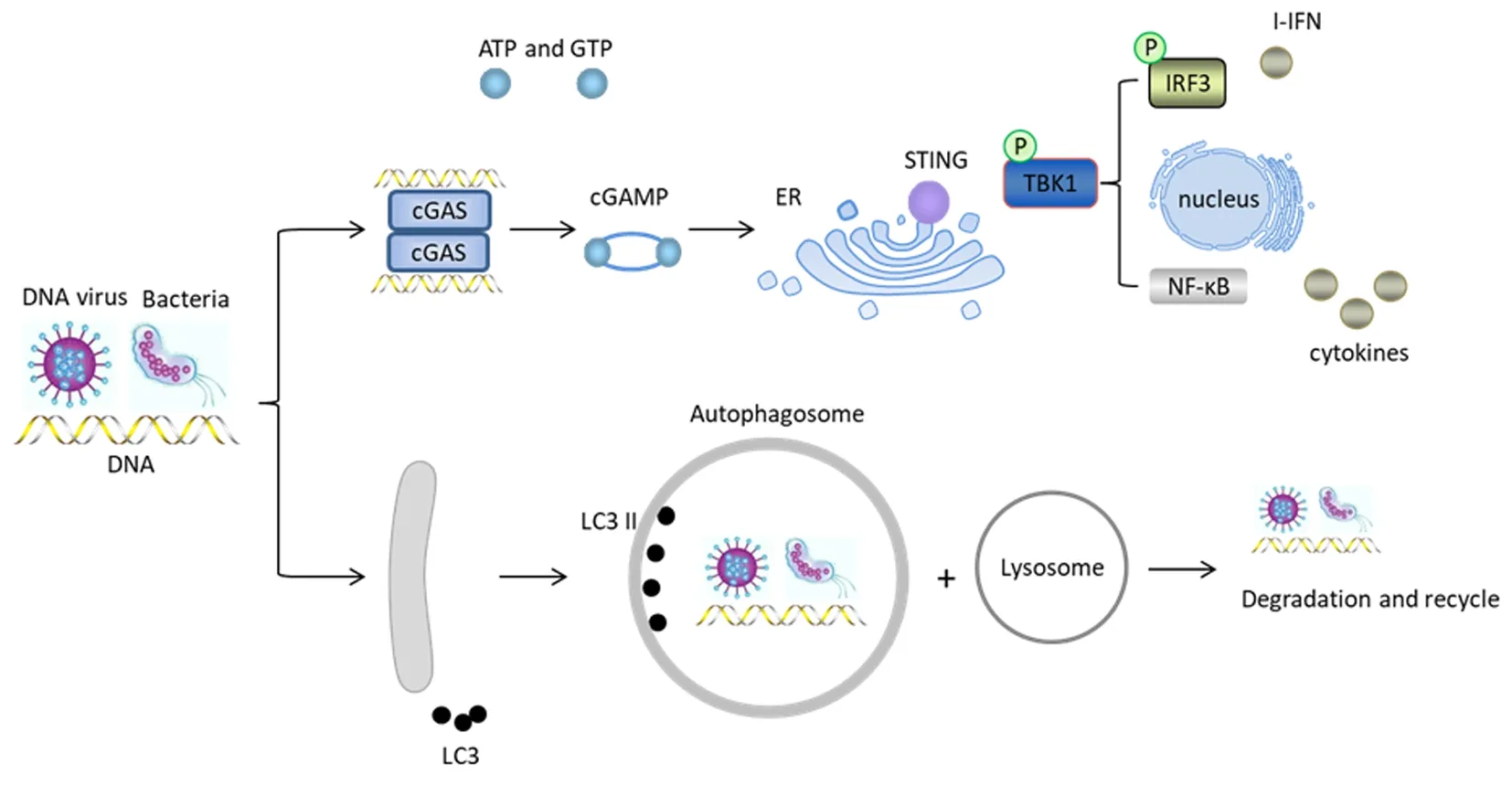
图 1 胞质dsDNA诱导cGAS-STING通路和自噬过程图
生理状态下,机体免疫反应能清除病原微生物而不造成组织损伤,这可能依赖于STING蛋白在免疫激活和自噬中的双重作用[5]。STING一方面通过cGAS-STING通路促进IFN、促炎因子的释放[2],激活免疫反应;另一方面活化的STING可与微管相关蛋白1轻链3 (LC3)相互作用启动自噬,致使STING被降解,防止免疫反应持续激活[5]。近年来大量研究证明cGAS-STING通路中各环节的信号分子与自噬相关基因(autophagy-related genes,ATG)存在交互作用,可诱导自噬,随后自噬通过降解cGAS-STING通路关键蛋白实现对该通路的负反馈调节[6]。
免疫过度激活与肠道炎症、自身免疫病等炎性疾病有关,自噬与 cGAS-STING 通路稳定的交互作用可减少免疫反应过度激活导致的组织损伤[5-6],故探究自噬与cGAS-STING 通路相互作用的具体机制对相关疾病的治疗有着重要的意义。本文就cGAS-STING通路与自噬的相互作用以及自噬相关蛋白对cGAS-STING通路的调控作用及其分子机制进行综述,以便为靶向cGAS-STING通路的抗感染治疗提供新思路。
1 STING通路上游信号与自噬的关联
cGAS在哺乳动物中具有DNA 感受器的功能,能识别胞质dsDNA并产生第二信使cGAMP,激活STING蛋白,启动固有免疫反应,是STING通路的上游信号分子[7]。结构上,cGAS由N-末端DNA结合结构域(DNA binding domain,DBD)、中心核苷酸转移酶(nucleotidyl transferase,NTase)结构域以及C-末端保守的Mab-21 (male abnormal 21)结构域组成,属于MAB21家族蛋白[7-9]。cGAS识别并结合dsDNA形成复合体,随后cGAS中心NTase结构域催化ATP和GTP合成cGAMP[10]。
Beclin 1是酵母Atg6的同系物,也是哺乳动物参与自噬的特异性基因[11]。 Beclin 1-PI3KC3 (Vps34)核心复合物通过生成富含磷脂酰肌醇3-磷酸(phosphatidylinositol 3-phosphate,PtdIns-3-P)的膜在诱导自噬过程中发挥关键作用,该膜可作为自噬蛋白募集和自噬体成核的平台[12]。大量研究表明,Beclin 1通过与其正(Atg14 和 UVRAG)或负(Bcl-2 和 Rubicon)调节蛋白形成各种复合物,在自噬诱导和自噬体成熟中发挥关键作用[13-15]。Rubicon与Beclin 1-PI3KC3 (Vps34) 核心复合物发生相互作用,抑制 PI3KC3 活性,从而在膜融合步骤负调节自噬[12]。
cGAS与Rubicon竞争性结合Beclin 1。cGAS的中心NTase结构域与Beclin 1的结合一方面抑制了cGAMP和IFN的合成;另一方面,该结合导致负自噬因子Rubicon从Beclin 1复合物上解离,使PI3KC3激活从而诱导自噬,清除胞质dsDNA,抑制cGAS-STING通路的持续激活。这表明在宿主免疫反应的后期,cGAS可能在STING介导的I型IFN途径和 Beclin 1介导的自噬途径之间穿梭,促进IFN表达的同时诱导自噬促进dsDNA降解以避免持续的免疫刺激。既往有研究表明,在二手烟暴露的条件下,Beclin 1缺乏可能导致小鼠线粒体DNA的释放和TNF-α等促炎因子的表达显著增加,最终引发心脏结构和功能缺陷,而cGAS-STING通路的抑制剂可减轻其炎症和心脏功能的障碍[21]。因此,胞质DNA传感器cGAS可通过Beclin 1协调IFN和自噬途径,最终优化机体炎症反应的时间和效率[11]。
2 STING蛋白与自噬的关联
cGAS-STING通路激活后可通过其各环节信号分子启动细胞的自噬机制,其关键蛋白STING本身也成为自噬降解的目标,这表明cGAS-STING通路与自噬存在依赖于STING蛋白的交互作用。
2.1STING直接诱导自噬有研究已揭示诱导自噬是cGAS-STING通路的一种原始功能[6]。自噬有非选择性和选择性之分。自噬的选择性由自噬受体决定,许多自噬受体的蛋白质序列包含LC3结合区域(LC3-interacting region,LIR),受体借助LIR与定位在自噬体上的LC3蛋白结合,将胞质内容物转运到自噬体中。LIR-LC3相互作用对于自噬体的形成、运输和成熟至关重要[16]。
作为固有免疫级联反应的关键调节因子,STING活性必须受到严格的调节,以确保适度的免疫反应,故自噬对STING的负调控可抑制机体过度的炎症反应。HSV-1感染可诱导细胞中LC3-II的转化和自噬流显著增加,采用氯喹抑制自噬流,宿主细胞中STING的蛋白含量增加,验证了自噬对STING的负调控[5]。
STING的二聚化和其与cGAMP的结合对于STING直接诱导的自噬至关重要[5]。结构分析表明,STING存在保守的LIR结构域,在激活时发生构象变化使LIR结构域暴露于胞质中,从而使其LIR结构域能够直接与 LC3 相互作用以激活自噬,导致 STING 本身和 p-TBK1 的降解。这种STING直接诱导的非典型自噬途径不依赖于 ULK1、Beclin1 和 Atg9a,但必须依赖于ATG5[5]。STING具有免疫激活和自噬诱导的双重作用:一方面STING激活后可促进I型IFN及其他炎症因子的产生;另一方面,STING 激动剂也可诱导 STING 依赖性自噬,从而对免疫反应进行调节,在防止持续的炎症反应造成严重的组织损伤方面发挥积极作用。
2.2ULK1磷酸化STING促进其被自噬降解Atg1/ULK1 (uncoordinated-51-like kinase)是一种丝氨酸/苏氨酸蛋白激酶,在自噬的多个阶段发挥作用。ULK1能够与Atg13、Atg101以及FIP200在胞质形成蛋白复合物,共同参与自噬启动和自噬体的形成。既往文献[12]报道,由腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)、哺乳动物雷帕霉素靶点 (mammalian target of rapamycin,mTOR)以及ULK1复合物共同组成AMPK-mTOR-ULK1通路,在调控自噬中起到重要作用。
AMPK和mTOR通过协调 ULK1 的磷酸化从而调节自噬[17]。在营养充足的条件下,mTOR通过磷酸化ULK1的丝氨酸758位点,抑制ULK1激酶复合物功能[18]。AMPK 是一种异源三聚体蛋白和关键的能量感应激酶,可被Ca2+外流或 ATP 消耗激活。在饥饿条件下,AMPK通过磷酸化直接激活ULK1来促进自噬[19-20]。此外,AMPK还能够通过磷酸化mTOR中的raptor蛋白抑制mTOR活性,进而诱导自噬[21]。因此,AMPK通过直接激活 ULK1,及抑制 mTOR活性间接诱导ULK1介导的自噬。
研究表明,cGAMP以STING非依赖的方式促进AMPK和ULK1活化,从而诱导自噬,ULK1激活后可磷酸化STING丝氨酸366位点(S366),该位点磷酸化后的STING蛋白通过自噬被降解,进而限制下游I型IFN效应[22]。因此,尽管cGAMP最初促进STING功能,但其随后会触发STING的负反馈调控,从而抑制促炎因子的表达。
类泛素蛋白修饰分子(small ubiquitin-like modifier,SUMO)是与泛素类似的蛋白之一,可介导SUMO化修饰过程,从而调控目的蛋白的定位和活性。既往文献报道,ULK1和TBK1介导STING在S366位点的磷酸化,促进其募集SUMO特异性蛋白酶家族2、去SUMO化和降解。在正常状态或感染早期,STING被E3泛素连接酶TRIM38 SUMO化修饰,促进其稳定性;而在感染后期,STING发生去SUMO化,进而促进STING的降解[23]。STING的稳定和降解对于感染和炎症的控制极为重要,成为调控机体炎症、感染状态和器官损伤的干预靶点。
2.3Atg9a调控STING与TBK1组装Atg9a是哺乳动物的多跨膜蛋白,也是一种自噬相关蛋白,位于高尔基体和晚期内体上,但并不固定于一个特定位点,而是在饥饿条件下于细胞器之间循环,调节膜转运以产生自噬体。在饥饿条件下,Atg9a被募集到自噬前体结构中,并作为隔离膜(IM)的初始膜来源[24]。Atg9a 蛋白的精确分子功能仍不清楚,但有研究提出 Atg9a 介导膜转运以产生自噬体,Atg9a与Atg2共同定位于隔离膜的扩展边缘,Atg2接收来自内质网的磷脂,由Atg2传递的磷脂通过Atg9a从细胞质转移到自噬体内膜小叶,从而驱动自噬体膜的扩张[24]。
Atg9a可调控STING介导的固有免疫反应。dsDNA刺激后,STING与自噬相关蛋白、LC3和Atg9a共定位, Atg9a 依赖性膜运输显著抑制了 STING 和 TBK1 的组装。Atg9a的缺失,极大地增强了dsDNA诱导的STING-TBK1复合体形成,导致固有免疫的异常激活[25]。因此,Atg9a作为dsDNA刺激后固有免疫的负调控因子发挥作用。
3 STING通路下游信号与自噬的关联
3.1 TBK1磷酸化P62 /SQSTM1促进自噬P62是选择性自噬受体大家族的一部分,该家族通过泛素结合结构域(ubiquitin binding domain,UBD)和LIR将泛素化与自噬联系起来。P62的表达与线粒体功能紊乱、ROS上调有关,当ROS浓度升高时,编码P62的SQSTM1基因可由转录因子Nrf2反式激活,P62与泛素化蛋白结合后,可通过其LIR结构域与LC3相互作用,靶向自噬体,促进其内容物快速降解[12]。
感染、组织损伤等内稳态异常诱导免疫激活,但STING蛋白的过度积累会引发自身免疫病和炎症性疾病[26]。故cGAS-STING途径激活后,一方面STING在E3泛素连接酶(如TRIM32、RNF26等)的作用下形成K63连接的泛素链[27-28];另一方面活化的TBK1使P62的丝氨酸403 位点磷酸化,增加了P62对 K63 连接的泛素链的亲和力[29-30],促进P62与STING的结合,从而使泛素化的STING靶向自噬体,抑制持续的免疫反应。因此,P62 /SQSTM1 介导的STING 降解依赖于 TBK1的激活。此外,E3 泛素连接酶 RNF26 和 TRIM30a 可促进 STING 发生 K48 连接的泛素化,从而使STING通过蛋白酶体途径降解[27, 31],因此泛素化的STING的降解并不局限于自噬途径。
3.2CALCOCO2/NDP52识别K27泛素化的IRF3除SQSTM1/P62外,CALCOCO2/NDP52(calcium binding and coiled-coil domain 2)、OPTN(optineurin)和 NBR1(next to BRCA1 gene 1 protein)也是选择性自噬受体,均含有UBD和 LIR结构域以确保将泛素化底物捕获到自噬体[32]。选择性自噬靶向一系列关键蛋白,如cGAS、MAVS 和 STING进行降解,从而负向调节固有免疫反应的激活。
IRF3是促进 I 型IFN表达的重要转录因子。生理情况下,去泛素化酶 PSMD14选择性地与非磷酸化的 IRF3 相关联,在 IRF3 上的赖氨酸 313 位点(K313)处切割 K27 连接的泛素链,防止 IRF3 发生自噬降解,从而维持IRF3的基础水平和 IRF3 介导的 I 型 IFN 表达[33]。病原微生物入侵时,在接收上游信号后,IRF3 经历多种过程以激活 I 型 IFN 信号传导,包括磷酸化、二聚化、易位到细胞核等[34]。另一方面,IRF3 复合物中的 PSMD14 释放,E3 连接酶(如 TRIM21)向 IRF3 复合物募集,在IRF3 K313位点诱导K27连接的泛素化。随后,自噬受体NDP52识别泛素化的IRF3靶向自噬体,进而减少I型 IFN 信号传导,避免下游细胞因子的过度表达[33]。
3.3I型IFN参与诱导自噬I型IFN是具有抗病毒、抗增殖和免疫调节等多种功能的蛋白质,是固有免疫反应的重要调节因子。IFN作用于靶细胞上的I型IFN受体(IFNAR),启动细胞内信号转导途径,导致抗病毒和免疫调节基因的表达,刺激各种免疫细胞。多项研究表明,I型IFN在多种细胞系中激活Janus激酶/信号转导和转录激活因子(Janus kinase/signal transducer and activator of transcription,JAK/STAT)和磷脂酰肌醇 3 激酶/蛋白激酶 B/哺乳动物雷帕霉素靶蛋白(phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin,PI3K/Akt/mTOR)信号途径参与自噬的诱导[35-38]。
IFN与其受体IFNAR1/2结合,通过酪氨酸激酶(TYK)2和JAK1诱导STAT1/2磷酸化。然后,磷酸化的STATs与IRF9结合,形成转录激活因子复合物ISGF3。随后ISGF3复合物转移至细胞核,与IFN刺激基因(ISG)的启动子区结合,促进其转录。有研究报道敲除JAK1或STAT1/2后,IFN无法促进LC3-II的转化,因此JAK/STAT 途径在IFN 诱导自噬中发挥重要作用[35, 39]。
IFN激活PI3K/AKT/mTOR途径需依赖于JAK1/TYK2,其磷酸化胰岛素受体底物1和2 (IRS1/2),导致PI3K激活,随后激活雷帕霉素靶蛋白复合物1(mTORC1), mTORC1激活的核糖体蛋白S6激酶(RPS6KB,也称为p70S6激酶)是IRS1的负调节因子。因此,I型IFN表达的中后期,IRS1的负反馈调节启动,PI3K/AKT/mTOR通路的负调节因子被激活,其抑制mTORC1活性并导致自噬的诱导[38]。此外,I型IFN还可激活雷帕霉素不敏感复合物(mTORC2), 从而活化AKT,AKT具有多个靶标,如FOXO3(Forkhead box O3),FOXO3也是自噬基因的直接转录调节因子[40]。因此,I型IFN激活mTORC2/AKT可能通过调节FOXO3诱导自噬。
4 结语与展望
cGAS-STING通路在固有免疫激活和自噬诱导中具有双重作用。病原微生物来源的 dsDNA 通过cGAS-STING-TBK1-IRF3/NF-κB轴促进I型IFN和促炎细胞因子的表达,增强免疫反应。另一方面,cGAS-STING通路上下游的多种调控因子和关键蛋白可与自噬相关蛋白相互作用,通过不同途径激活自噬通路,导致cGAS-STING通路自身各环节的降解。故cGAS-STING通路直接将固有免疫激活与自噬诱导结合,从而限制免疫反应的过度激活,减少自身组织损伤。
减少免疫反应造成的组织损伤是改善感染或炎性疾病预后的关键,目前已有许多研究致力于探究基于调节自噬的相关炎症疾病疗法,如自噬诱导剂雷帕霉素类似物依维莫司的治疗可改善IL-10缺陷小鼠的回肠炎症状。然而,雷帕霉素类似物的有益作用也可能是由与自噬无关的免疫抑制作用产生的。自噬调节剂作为一种疗法的使用仍然具有挑战性,因为其靶标的药理学特异性低,存在与自噬无关的效应,缺乏对特定细胞类型的特异性效应。故无毒性作用的特异性调节自噬的药物仍需要深入研究,使其最终可用于临床相关炎症疾病的治疗。因此对自噬调节cGAS-STING通路的相关机制的探究能够为相关临床疾病的治疗提供新的方向。
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
中国药理学与毒理学杂志(2022年7期)2022-10-17
生物化学与生物物理进展(2022年8期)2022-08-20
九江学院学报(自然科学版)(2022年2期)2022-07-02
波谱学杂志(2022年1期)2022-03-15
中草药(2022年5期)2022-03-03
昆明医科大学学报(2022年1期)2022-02-28
实用肿瘤学杂志(2021年3期)2021-11-29
心血管病学进展(2021年8期)2021-09-13
中华骨与关节外科杂志(2021年12期)2021-08-31
