
7S大豆球蛋白α-亚基的原核表达及分离纯化
2022-05-18 09:10:34高梦楠袁艳秋胡亚云栾广忠
西北农业学报 2022年1期
高梦楠,袁艳秋,巨 倩,胡亚云,栾广忠
(西北农林科技大学 食品科学与工程学院,粮油功能化加工陕西省高校工程研究中心,陕西杨凌 712100)
大豆蛋白主要由7S(35%)与11S(52%)大豆球蛋白构成[1],其中,7S大豆球蛋白(β-伴大豆球蛋白,β-conglycinin)对大豆蛋白功能性质有很大影响[2]。β-伴大豆球蛋白是一种复合糖蛋白,分子质量为150~200ku,由α (≈67ku),α′ (≈71ku),β (≈50ku)通过非共价键以不同的组合形成三聚体[3-5]。目前分离纯化7S大豆球蛋白3个亚基难度较大,且无法大量制备,制约了研究的深入[8-11]。植物蛋白课题组前期通过基因重组技术完成重组大豆7S球蛋白α′-亚基及α-亚基核心区的基因克隆、原核表达及分离纯化[6-8],得到足够量的亚基(或片段)材料,有效解决了α′-亚基及α-亚基核心区试验材料的制备问题。
本研究对β-伴大豆球蛋白的α-亚基进行基因克隆、表达及分离纯化,以期为后续构建大豆蛋白水分散体系[13]提供α-亚基材料。
1 材料与方法
1.1 试验材料
‘齐黄34’大豆,2019年种植并收获于山东省济南市,山东省农业科学院提供;感受态细胞E.coliDH5α、E.coliBL21(DE3)、质粒pET-28a(+),西安热默尔生物科技有限公司提供;质粒pGEM-T Easy,普洛麦格(北京)生物技术有限公司提供。
1.2 主要试剂与仪器
PrimeScriptTMⅡ 1st Strand cDNA Synthesis Kit、QuickCutTMEcoRⅠ、QuickCutTMXhoⅠ、DL 5000 DNA Marker、Premixed Protein Maker (Broad),宝生物工程(大连)有限公司,中国。异丙基硫代-β-D-半乳糖苷(IPTG),北京索莱宝有限公司,中国;氨苄西林钠盐(Amp)、硫酸卡纳霉素(Kana),北京拜尔迪生物技术有限公司,中国;其余试剂均为国产分析纯。
JY-SCZ2+电泳仪,北京君意东方电泳设备有限公司,中国;JY92-IIDN 超声波细胞粉碎机,新芝(浙江宁波)生物科技有限公司,中国;ClearFirst-2000型蛋白纯化系统,闪谱(上海)生物科技有限公司,中国。
1.3 试验方法
1.3.1 重组克隆载体、表达载体的构建 根据NCBI官网(https://www.ncbi.nlm.nih.gov/)α-亚基编码的基因序列(参考序列号为NM_001249927.2)设计一对特异性引物。上游引物(5′-GCGGACGACGACGACAAGGTGGAGAA- AGAAGAAT-3′)引入保护碱基和肠激酶酶切位点(加粗部分)方便后期去除纯化后的重组α-亚基蛋白N-端标签;下游引物(5′- ATACCGCTCGAGTTCAGTAAAAAGCCCTCAA-3′)引入XhoⅠ酶切位点(加粗的6个碱基)。
参照李垚熹等[12]的方法获得α-亚基的cDNA,利用Premix Taq酶扩增α-亚基,设置模板缺失对照。将理论值约为1 660 bp的PCR产物,利用T4-DNA连接酶与线性化的克隆载体pGEM-T Easy相连接,构建pGEM-T Easy-α,并导入感受态细胞E.coliDH 5α中[12]。pGEM-T Easy-α与pET-28a表达系统同时利用XhoⅠ和EcoRⅠ酶切出相同的粘性末端[14],通过T4-DNA连接酶连接构建pET-28a-α,并导入感受态细胞E.coliBL21(DE3)中。构建结果分别经蓝白斑筛选、菌落PCR筛选[7]、EcoRⅠ/XhoⅠ单双酶切鉴定[15-16],对插入的α-亚基区域外送测序鉴定,测序结果通过NCBI进行BLAST分析,以确保所构建的pGEM-TEasy-α、pET-28a-α序列无误。命名测序正确的pET-28a-α阳性菌株为pET-28a-α-BL21。
1.3.2 重组蛋白α-亚基表达条件的筛选 将pET-28a-α-BL21增菌培养8 h后按φ=1%接种量接种至含有卡那霉素(50 μg/mL)的LB液体培养基中,培养至不同菌液浓度(OD600值为0.2、0.4、0.6、0.8),加入诱导剂IPTG(0.2 mmol/L)后采用不同诱导温度(25 ℃、30 ℃、 37 ℃),不同诱导表达时间(3 h、5 h、7 h、9 h、11 h)来筛选重组α-亚基蛋白的表达条件[17-18]。采用φ=10%分离胶和φ=5%浓缩胶进行SDS-PAGE检测,利用Image J软件进行条带灰度定性分析,目的条带灰度占全部条带灰度的百分数,即为目的蛋白占全蛋白的表达百分数。数据采用DPS 7.05进行单因素方差分析(ANOVA),结果以“平均数±标准差”表示。采用邓肯检验确定在0.05水平上的显著性差异。
1.3.3 重组蛋白的表达与提取 挑pET-28a-α-BL21单菌落转接至500 mL LB液体培养基(含卡那霉素50 μg/mL),利用“1.3.2”筛选出的条件诱导培养转化菌,同时设置不含α-亚基的pET-28a和不进行诱导表达的pET-28a-α(IPTG浓度为0 mmol/L)分别做对照[16]。转化菌液以7 500 r/min冷冻离心20 min,去除上层培养基得到菌体沉淀。添加60 mL磷酸盐缓冲液PBS(20 mmol/L磷酸钠缓冲液,0.5 mol/L NaCl,1 mmol/L EDTA,0.1 mmol/L PMSF,0.02% NaN3,pH=7.4)于菌体沉淀,漩涡震荡得到菌悬液,重复离心至上清清澈[12]。60 mL菌悬液超声参数为:总时间80 min、超声4 s、间隔6 s、功率270 W、12~15 ℃。超声破碎1次后将破碎菌体在4 ℃的环境中以8 500 r/min的转速离心45 min,取其上清液为目的蛋白α-亚基的粗蛋白液,用SDS-PAGE检测上清中表达的目的蛋白,其余在4 ℃环境中保存。
1.3.4 重组蛋白的分离纯化 提取的50 mL重组蛋白粗蛋白液使用0.45 μm孔径的水性滤膜过滤除杂。将5 mL的His Trap HP预装柱[20]安装于蛋白纯化系统,使用超声后的超纯水冲洗预装柱5~10个柱体积,接着使用超声后的平衡磷酸盐缓冲液(20 mmol/L磷酸钠缓冲液,0.5 mol/L NaCl, 1 mmol/L EDTA,0.1 mmol/L PMSF,0.02% NaN3,pH=7.4)平衡预装柱5~10个柱体积,待基线保持平衡使用自动上样泵完成上样。用超声后的平衡缓冲液(分别含有0 mmol/L、500 mmol/L咪唑)进行线性洗脱,确定目的蛋白出峰时平衡缓冲液咪唑浓度,收集流动相。用出峰咪唑浓度、500 mmol/L咪唑平衡缓冲液进行分步洗脱,收集不同阶段的流动相进行SDS-PAGE电泳检测鉴定。
2 结果与分析
2.1 重组克隆载体pGEM-T Easy-α的构建及鉴定
以所提取的大豆总RNA为模板,通过设计的上、下游引物RT-PCR扩增得到α-亚基基因 (1 660 bp),结果如图1-A所示,泳道1为引物结合位点之间的片段。
蓝白斑筛选出5个白色重组克隆载体pGEM-T Easy-α的单克隆菌株进行菌落PCR,结果如图1-B所示。泳道2、8、10得到的片段长度与α-亚基理论分子量(1 660 bp)相一致;泳道1、7、9得到的片段长度与通用引物PCR所得基因理论长度(2 000 bp)相符。因此,选用泳道1、2、7、8、9、10的3个菌株进行后续试验[19]。
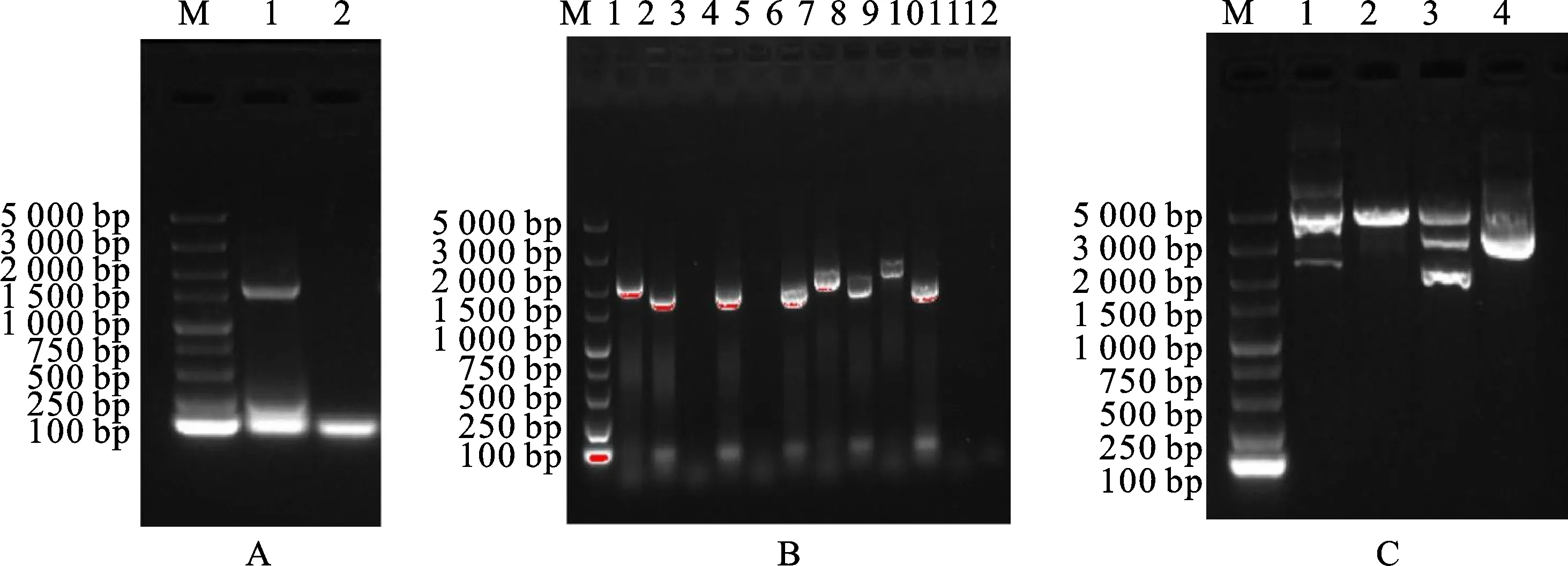
M. DNA marker;A. 目的基因的扩增;1. α-亚基基因扩增结果;2. 阴性对照;B.菌落PCR鉴定;1、3、5、7、9. 单克隆菌株使用M47/M48通用引物扩增;2、4、6、8、10. 单克隆菌株使用设计的上、下游引物扩增;11. 空白使用通用引物M47/M48进行扩增;12. 空白使用设计的上、下游引物进行扩增;C. 酶切鉴定pGEM-T Easy-α重组质粒;1~3 .重组质粒pGEM-T Easy-α分别经EcoR Ⅰ、Xho Ⅰ酶单酶切、双酶切;4. 重组质粒pGEM-T Easy-α
酶切法鉴定重组克隆载体pGEM-T Easy-α结果见图1-C。泳道1为经EcoRⅠ单酶切形成两个清晰条带,是由于目的基因片段两端均带有EcoRⅠ酶切位点;泳道2为经XhoⅠ单酶切形成单一的条带(5 000 bp左右),与载体pGEM-T Easy(3 015 bp)和目的基因α-亚基(1 660 bp)成功相连后的理论值大小相符。泳道3为经EcoRⅠ、XhoⅠ双酶切得到5 000 bp、3 000 bp与 1 700 bp左右的条带,分别与目的基因与载体相连、载体、目的基因的理论值大小相符。
于NCBI进行BLAST比对分析,测序的 1 635个碱基序列与α-亚基编码区的243-1874碱基100%一致,表明构建序列无突变、移码,目的基因已正确插入到重组质粒pGEM-T Easy-α中。
2.2 重组表达载体pET-28a-α的构建及鉴定
图2-A、图2-B为随机挑选的6个pET-28a-α-BL21进行菌落PCR筛选的结果,可以看到泳道6、10、14、19、22扩增所得产物条带大小均在 1 500 ~2 000 bp,约为1 700 bp,与目的基因理论长度相符,考虑到条带的清晰度、弥散程度和亮度,选用泳道6、19的样品进行下一步试验。
如图2-C,泳道1中经EcoRⅠ单酶切得到 7 000 bp左右的条带,与pET-28a(5 369 bp)、 α-亚基基因(1 660 bp)成功相连接后的理论值相符,大于7 000 bp的条带应为未被酶切的pET-28a-α;泳道2中经XhoⅠ单酶切得到与线性pET-28a-α(7 029 bp)理论值相符的条带;泳道3经双酶切所得条带分子量大小正确(1 660 bp、5 369 bp)。
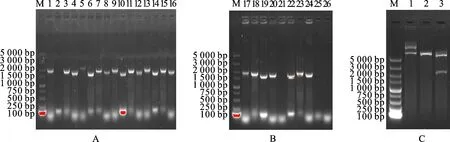
A、B. 菌落PCR鉴定;M. DNA marker;1、5、9、13、17、21. 单克隆菌株使用通用引物T7/T7Ter进行扩增;2、6、10、14、19、22. 单克隆菌株使用设计的上、下游引物进行扩增;3、7、11、15、18、23. 单克隆菌株使用T7和设计的下游引物进行扩增;4、8、12、16、20、24. 单克隆菌株使用设计的上游引物和T7Ter进行扩增;25、26. 空白分别使用T7/T7Ter和设计的上、下游引物进行扩增;C. 酶切鉴定重组质粒pET-28a-α;M. DNA marker;1-3. 分别经EcoRⅠ、XhoⅠ酶单双酶切使pET-28a-α线性化
于NCBI进行BLAST比对分析,测序的 1 666个碱基序列与α-亚基编码区的243-1874碱基100%一致,表明构建序列无突变、移码,α-亚基基因已正确插入到重组表达载体pET-28a-α。
2.3 重组蛋白表达条件的筛选
图3泳道1~20表明重组α-亚基蛋白分子质量约70 ku,单独携带pET-28a的阴性对照未检测到目的蛋白带(图3泳道21)。利用Image J软件对图3条带进行灰度分析,结果如表1,目的蛋白表达含量小于10%的未列出。图3-E、图3-F条带浅、较为弥散,表明在25 ℃大肠杆菌全蛋白表达含量较少,虽然泳道17目的蛋白表达占比例较高,但其含量不理想;图3-A、图3-B条带较深,表明37 ℃适宜大肠杆菌进行蛋白表达,但是重组α-亚基蛋白表达占比例较低。结合表1数据,筛选出高效表达靶蛋白条件为菌液浓度(OD600) 0.6,30 ℃诱导培养9 h。此条件下目的蛋白表达量占全菌蛋白的25.83%。
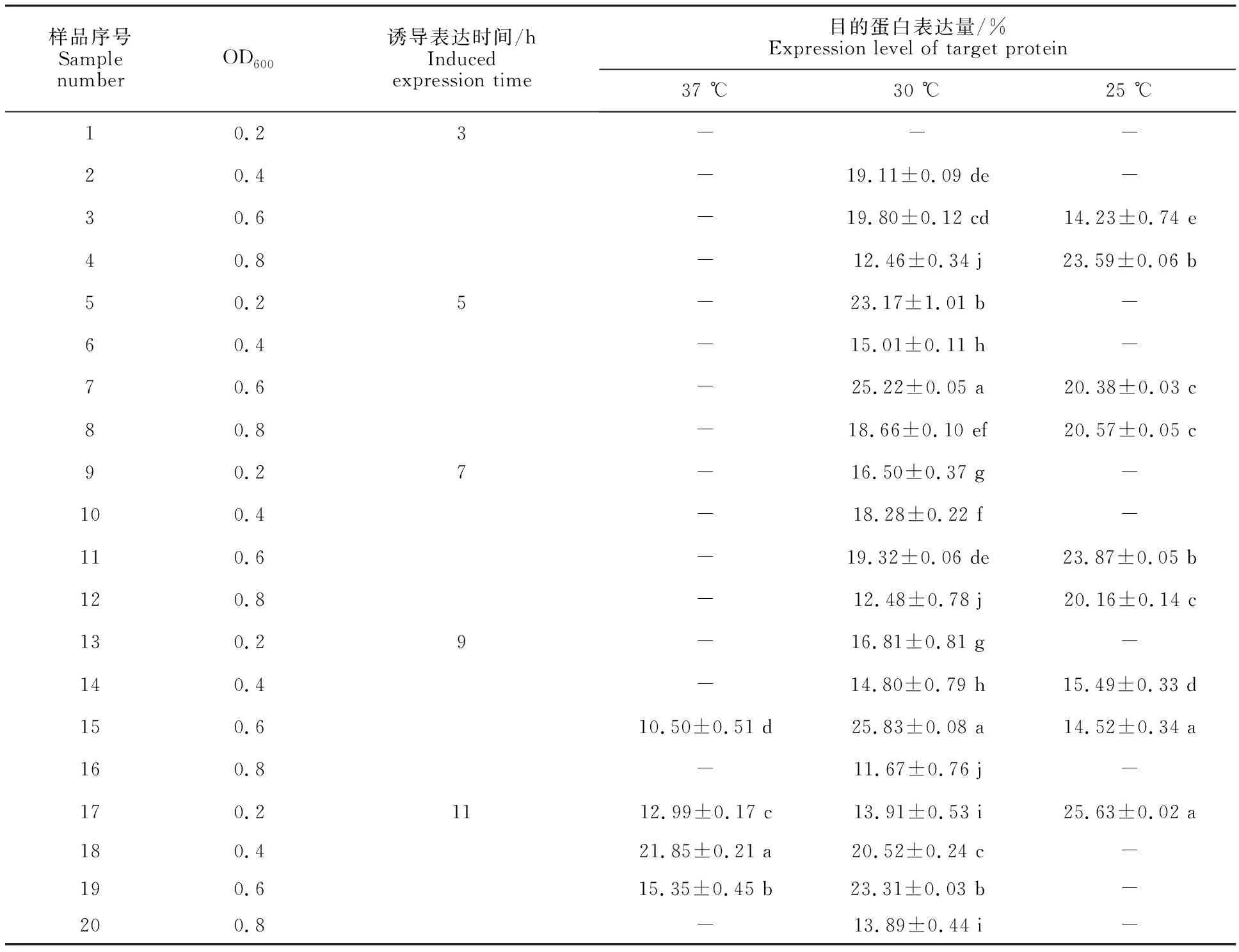
表1 重组α-亚基蛋白的表达量Table 1 Expression of recombinant α-subunit protein
2.4 重组α-亚基在大肠杆菌中的表达及分离纯化
利用“2.3”筛选出的条件培养pET-28a-α-BL21,超声波破碎大肠杆菌释放出靶蛋白的结果如图4所示。与两个对照(图4泳道3、4)相比较,泳道1、2中均出现分子质量在70 ku左右的目的蛋白条带,由于上清液中含有具有较好溶解性的目的蛋白(未形成包涵体)[21-23],因此泳道2样品可以进行重组蛋白的分离纯化。
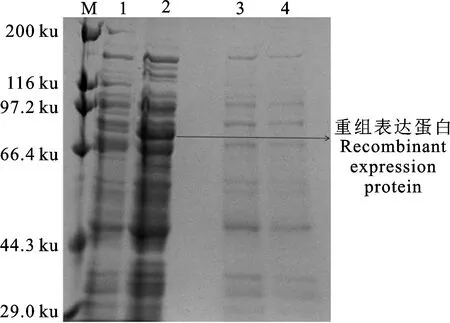
M. 蛋白标marker;1~2. pET-28a-α诱导培养后的全菌液、上清液;3. pET-28a-α经破碎后的上清液;4. 表达载体pET-28a
用His结合树脂对泳道2的样品进行镍亲和层析纯化。两次泵入10 mL样品进行线性洗脱结果如图5,洗脱液体积分数为70%(350 mmol/L的咪唑平衡缓冲液)时出现洗脱峰。将上样峰(F1)与洗脱峰(F2)使用超滤离心管浓缩后的溶液及滤液进行SDS-PAGE电泳检测,结果如图6-A,目的蛋白在洗脱峰F2中的含量较高(泳道4),但由于存在杂带,因此决定使用分步洗脱(选用350、500 mmol/L的咪唑平衡缓冲液)进一步纯化目的蛋白。

F1. 蛋白纯化上样峰;F2. 蛋白纯化洗脱峰

M.蛋白质marker;A. 线性洗脱结果;B.分步洗脱结果;A:1. 未纯化的样品;2. 蛋白纯化F1峰;3~4. 蛋白纯化F2峰浓缩滤液、浓缩液;B:1. 上样回收液;2~4 纯化后样品(分别用0 mmol/L、500 mmol/L和350 mmol/L咪唑的平衡缓冲液洗脱)
分步洗脱峰的流动相经SDS-PAGE检测鉴定所得结果如图6-B。泳道1存在分子质量70 ku的重组α-亚基蛋白带,因为部分目标蛋白未结合到Ni2+亲和柱上,随平衡缓冲液流出;泳道2未发现有目的蛋白条带,表明重组蛋白通过His标签和亲和柱上的镍离子相结合,留在柱材上,未被平衡缓冲液洗脱下来;泳道4经Image J软件进行灰度分析,所得目标蛋白纯度可达到70.0%左右。
3 结 论
成功构建重组表达载体pET-28a-α并导入E.coliBL21(DE3)中。筛选出高效表达重组7S大豆球蛋白α-亚基的条件为:菌液浓度(OD600) 0.6,诱导培养温度30 ℃,时间9 h。此条件表达的目的蛋白达大肠杆菌全菌蛋白的25.83%。利用镍亲和层析柱,通过350 mmol/L的咪唑磷酸盐缓冲液进行洗脱,最终获得纯度可达70.0%左右的目的蛋白。
猜你喜欢
睿士(2023年9期)2023-09-20 05:47:07
世界最新医学信息文摘(2020年68期)2020-12-25 11:55:27
生物工程学报(2019年1期)2019-01-30 08:19:58
小雪花·初中高分作文(2016年9期)2016-05-14 02:50:08
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
中国光学(2015年1期)2015-06-06 18:30:20
海岸工程(2014年4期)2014-02-27 12:51:28
中国医学科学院学报(2013年6期)2013-03-11 20:26:01
中国烟草学报(2012年5期)2012-04-12 06:21:19
