
长链非编码RNA CBR3-AS1通过PI3K/AKT/mTOR/S6K通路介导白血病细胞对阿糖胞苷的耐药机制
2022-05-17 04:21:28李晓明郑丽飞
实用临床医药杂志 2022年8期
高 莉, 李晓明, 杨 波, 秦 英, 程 冬, 郑丽飞, 李 里
(1. 四川省雅安市人民医院 血液内科, 四川 雅安, 625000;2. 西南医科大学附属医院 血液内科, 四川 泸州, 646000; 3. 四川省南充市中心医院, 四川 南充, 737003)
急性髓细胞白血病(AML)是一类常见的造血干细胞恶性克隆性疾病,以阿糖胞苷为代表的核苷类似物是常用的治疗AML的化疗药物[1-2]。和许多其他化疗药物一样,阿糖胞苷等也不可避免地出现耐药情况,导致AML的完全缓解率不足50%,且容易产生耐药[3-4]。目前关于阿糖胞苷的耐药机制仍不十分清楚,致使临床上在应对阿糖胞苷耐药时的干预策略有限,限制了AML患者治疗疗效。
长链非编码RNA(lncRNA)是指一类长度200~100 000 nt的RNA分子,在包括AML在内的多种恶性肿瘤的发生发展过程中具有重要作用[5-6]。lncRNA CBR3-AS1是近年来研究较热的lncRNA家族成员,如lncRNA CBR3-AS1被报道[7]在乳腺癌中高表达,且和TNM分期、淋巴结转移、远处转移等相关,和更低的无瘤生存率、总生存率相关; lncCBR3-AS1促进了骨肉瘤细胞的增殖、迁移及侵袭能力,且是影响骨肉瘤预后的独立危险因素[8]。研究[9-10]报道lncRNA参与了AML的阿糖胞苷耐药过程,但lncRNA CBR3-AS1在AML中的作用及其与化疗耐药的关系的报道较少。本研究发现lncRNA CBR3-AS1在阿糖胞苷耐药的AML细胞株中表达升高,且lncRNA CBR3-AS1通过调控PI3K/AKT/mTOR/S6K通路调节阿糖胞苷的耐药,现报告如下。
1 材料与方法
1.1 细胞系的培养与耐药株的构建
AML细胞系K562(人髓系白血病细胞株)和HL-60(人急性早幼粒白血病细胞)均购自上海中科院细胞库,采用含10% 胎牛血清的RPMI 1640培养液悬浮培养, 37 ℃、5% CO2于培养箱中培养,每2~3 d传代1 次。构建耐药株时,以半数抑制浓度(IC50)为诱导的起始浓度,培养至细胞可稳定增殖后,以2倍浓度的浓度梯度递增,直至阿糖胞苷浓度超过1 000 μmol/L时细胞仍可稳定增殖,视为阿糖胞苷耐药。
1.2 IC50测定
对数生长期的待测细胞种板至96孔板中,设置8个浓度梯度,分别为5、1.5、0.45、0.135、0.040 5、0.012 15、0.003 645、0.001 093 5、0.000 328 05 mmol/L, 各设置8个复孔,培养24 h后每孔加入10 μL CCK8试剂,37 ℃孵育30 min, 在酶标仪450 nm波长处测定光密度(OD)值。细胞活性=(实验OD450 nm-空白OD450 nm)/(对照OD450 nm-空白OD450 nm)×100%。利用GraphPad软件计算IC50。
1.3 过表达质粒及siRNA的转染
构建lncRNA CBR3-AS1过表达及对照空载质粒(吉凯基因公司)、靶向lncRNA CBR3-AS1的siRNA及对照无靶向siRNA。细胞种至6孔板后,利用Lipo 3000转染质粒和siRNA, 转染48 h后,进行后续实验。靶向lncRNA CBR3-AS1 的siRNA序列为5′-GTCTCCTGAGCTCAGGAAA-3′, 对照siRNA序列为5′-GATATGGGCTGAATACAAA-3′。
1.4 逆转录-实时定量聚合酶链反应(RT-qPCR)检测lncRNA CBR3-AS1表达
取实验细胞,利用Trizol提取总RNA, 利用Genecopia逆转录试剂盒逆转录为cDNA, 依据定量PCR试剂盒说明书,进行实时荧光定量PCR。LncRNA CBR3-AS1的引物序列如下(5′→3′): 上游引物CTGTCGCCCAGGCTGGAGTGC; 下游引物GACGCCGTGGGTCCTTCTCATC。内参β-actin的引物序列如下: 上游引物CATGTACGTTGCTATCCAGGC; 下游引物CTCCTTAATGTCACGCACGAT。
1.5 蛋白印迹法
利用RIPA提取转染后的细胞总蛋白,加苯甲基磺酰氟(PMSF)和磷酸酶抑制剂防止蛋白降解与去磷酸化。一抗anti-p-PI3K、anti-p-AKT、anti-p-mTOR、anti-p-S6K、anti-GAPDH均购自Cell Signaling Technology, 二抗anti-mouse(1∶5 000)、anti-rabbit(1∶5 000)购自Affinity。总蛋白80 V恒压电泳后200 mA恒流转膜2 h, 5%脱脂奶粉室温封闭2 h, 一抗4 ℃摇床孵育过夜,二抗室温摇床孵育2 h后,用辣根过氧化物酶法检测蛋白表达。GAPDH作为蛋白印迹实验的内参。
1.6 统计学分析
采用SPSS 25.0软件包进行数据统计,计量资料采用均数±标准差表示,采用单因素方差分析或独立样本t检验进行比较,组间两两比较采用q检验,检验水平α=0.05。P<0.05为差异有统计学意义。
2 结 果
2.1 lncRNA CBR3-AS1在阿糖胞苷耐药的细胞株中表达升高
为了研究阿糖胞苷在AML细胞株中的耐药机制,建立了2个AML细胞株耐阿糖胞苷模型。首先检测K562和HL-60细胞株阿糖胞苷的IC50, 结果表明K562和HL-60的阿糖胞苷IC50分别为13.65 μmol/L和4.62 μmol/L。利用小剂量梯度递增法,建立了K562和HL-60耐阿糖胞苷的细胞株,分别命名为K562-R和HL-60-R, 2个耐药细胞株的IC50均超1 000 μmol/L(P<0.05)(图1A)。为了研究lncRNA CBR3-AS1在阿糖胞苷耐药中的作用, RT-qPCR检测lncRNA CBR3-AS1在野生及耐药细胞株中的表达,发现在K562-R和HL-60-R中, lncRNA CBR3-AS1的表达均升高,差异有统计学意义(P<0.05)(图1B), 提示lncRNA CBR3-AS1可能参与AML细胞阿糖胞苷耐药。
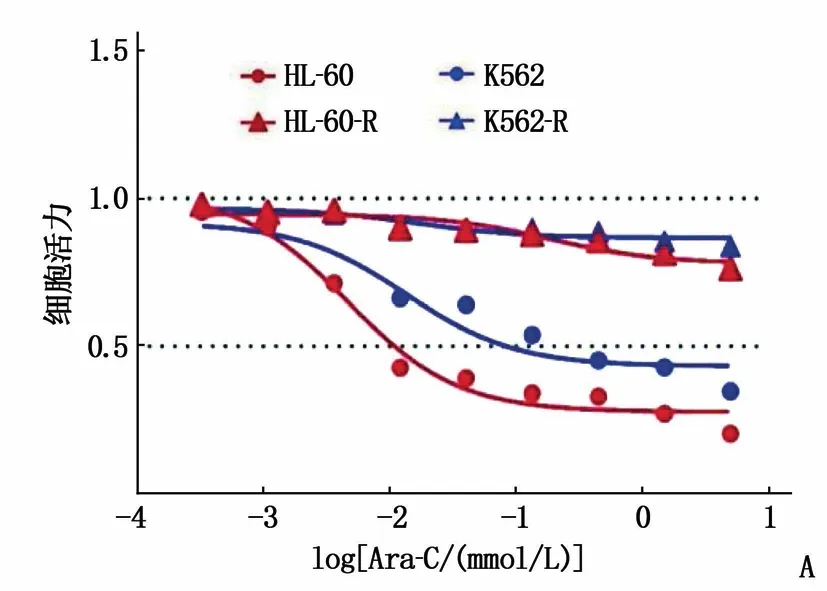
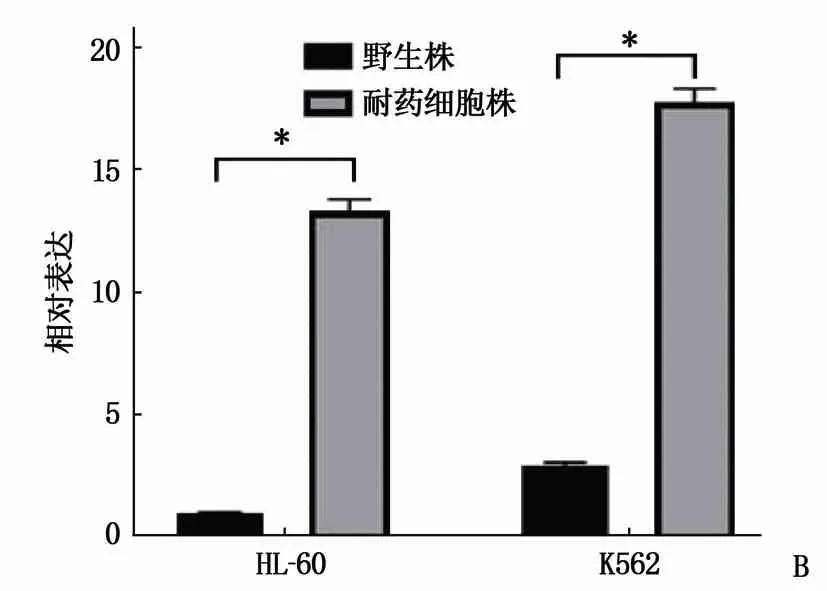
A: K562-R和HL-60-R对阿糖胞苷的耐药性明显增强; B: RT-qPCR结果提示K562-R和HL-6-R中,lncRNA CBR3-AS1的表达均升高,与野生株比较, *P<0.05。
2.2 过表达lncRNA CBR3-AS1导致野生株继发耐药
为验证lncRNA CBR3-AS1在AML细胞阿糖胞苷耐药中的作用,构建lncRNA CBR3-AS1过表达质粒并转染到K562和HL-60细胞株中(图2A), 利用CCK-8法检测IC50, 发现过表达lncRNA CBR3-AS1后,阿糖胞苷在K562和HL-60细胞的IC50升高并超过1 000 μmol/L, 差异有统计学意义(P<0.05)(图2B)。
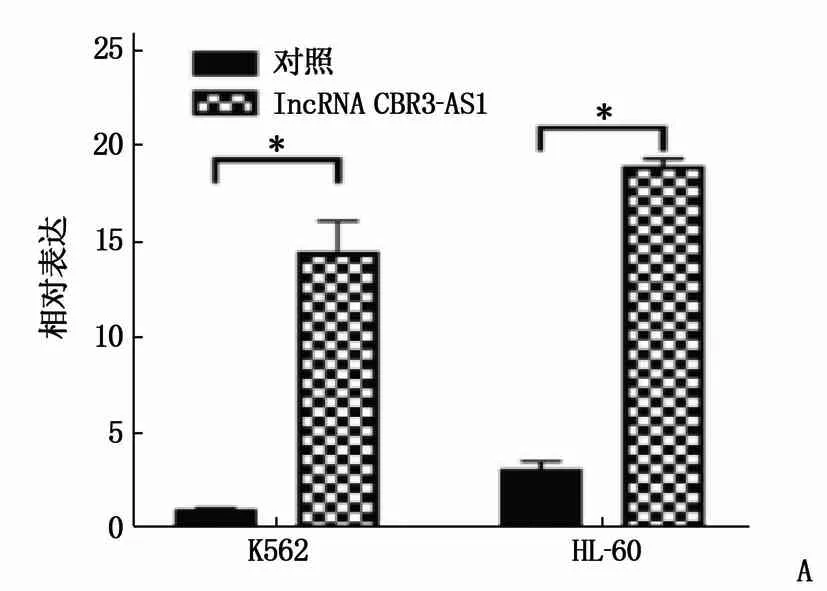

A: RT-qPCR验证lncRNA CBR3-AS1过表达质粒在K562和HL-60细胞株中的构建,与对照比较, *P<0.05;B: 过表达lncRNA CBR3-AS1后,阿糖胞苷在K562和HL-60细胞的IC50升高。
构建靶向lncRNA CBR3-AS1的siRNA, 并转染到K562-R和HL-60-R细胞系中(图3A)。利用CCK-8法检测IC50发现,敲低lncRNA CBR3-AS1后,阿糖胞苷在耐药细胞株K562-R和HL-60-R中的IC50分别降低到21.27 μmol/L和12.10 μmol/L, 差异有统计学意义(P<0.05)(图3B)。这些结果表明, lncRNA CBR3-AS1可能介导了AML细胞株的阿糖胞苷耐药。
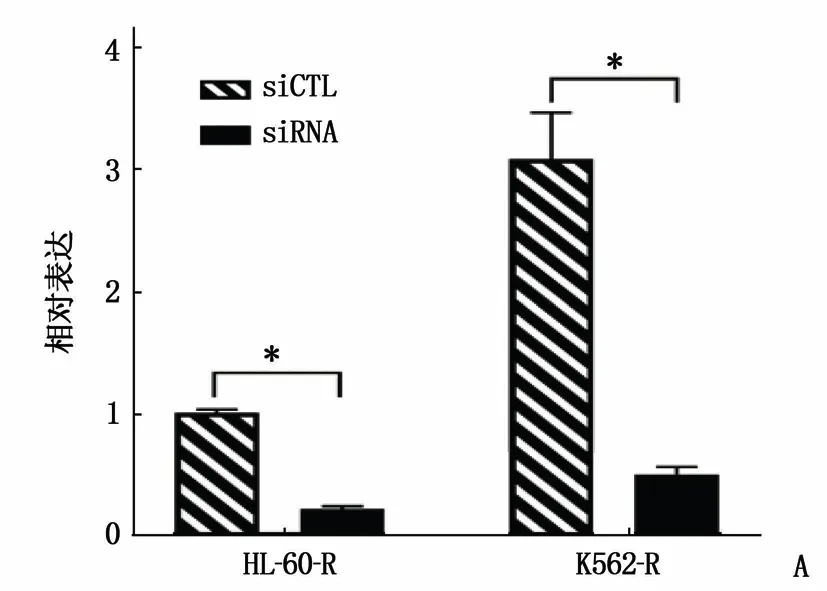

A: RT-qPCR验证siRNA lncRNA CBR3-AS1在K562-R和HL-60-R细胞株中的构建,与对照siCTL比较, *P<0.05;B: 敲低LncRNA CBR3-AS1后,阿糖胞苷在K562-R和HL-60-R细胞株中的IC50升高。
2.3 LncRNA CBR3-AS1通过调节PI3K/AKT/mTOR/S6K通路介导阿糖胞苷耐药
在K562-R细胞株过表达lncRNA CBR3-AS1, 蛋白印迹法检测发现,相较于对照(control), 过表达lncRNA CBR3-AS1提高了磷酸肌醇3激酶(PI3K)、AKT、哺乳动物雷帕霉素靶蛋白(mTOR)、S6K等蛋白的磷酸化水平,差异有统计学意义(P<0.05)(图4A)。在K562-R细胞株中,相较于对照中(siCTL), 利用siRNA敲低lncRNA CBR3-AS1, 降低了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平,差异有统计学意义(P<0.05)(图4B)。上述研究结果表明, lncRNA CBR3-AS1通过激活PI3K/AKT/mTOR/S6K通路介导了阿糖胞苷耐药。

A: 相较于control, 过表达lncRNA CBR3-AS1显著提高了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平; B: 相较于siCTL, siRNA中PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平显著降低。图4 lncRNA CBR3-AS1激活K562-R细胞株的PI3K/AKT/mTOR/S6K通路
在HL-60细胞株过表达lnc CBR3-AS1, 蛋白印迹法检测发现,相较于对照(control), 过表达lncRNA CBR3-AS1提高了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平,差异有统计学意义(P<0.05)(图5A)。在HL-60细胞株中,相较于对照(siCTL), 利用siRNA敲低lncRNA CBR3-AS1, 降低了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平,差异有统计学意义(P<0.05)(图5B)。上述研究结果表明, lncRNA CBR3-AS1通过激活PI3K/AKT/mTOR/S6K通路介导了阿糖胞苷耐药。
3 讨 论
目前, AML患者化疗的完全缓解率及无进展生存率仍较低,对于AML阿糖胞苷耐药的分子机制的研究将有助于进一步深入理解AML化疗耐药的分子生物学基础,对于临床开发新的克服耐药策略具有重要的指导意义。
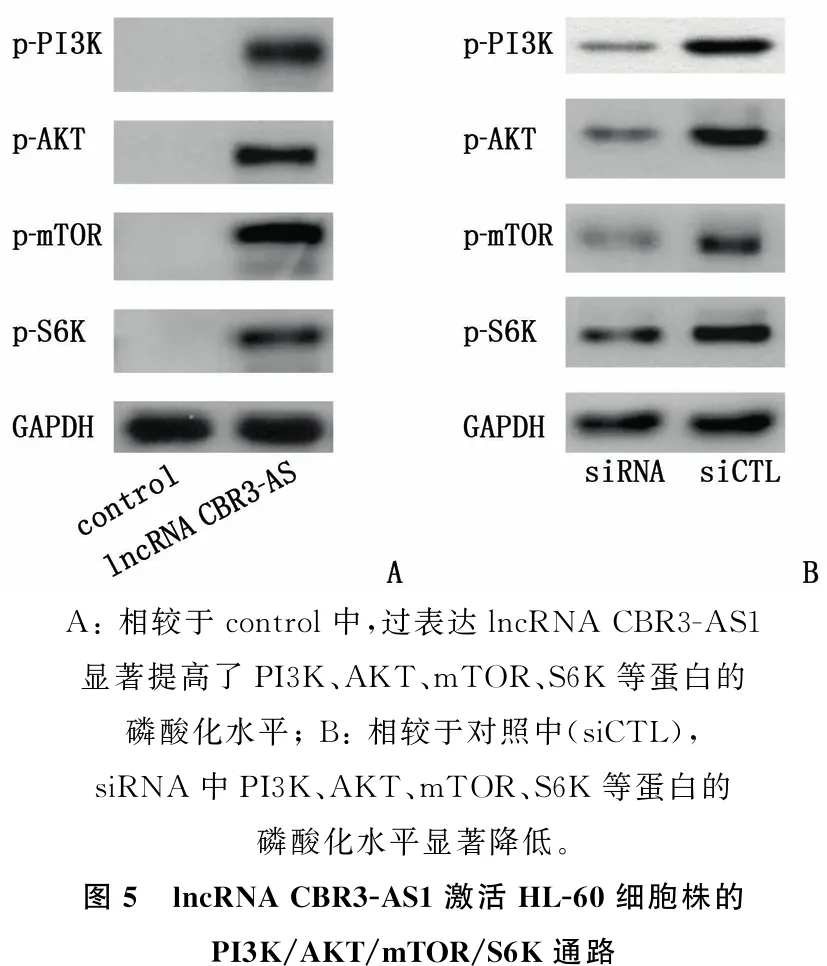
A: 相较于control中,过表达lncRNA CBR3-AS1显著提高了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平; B: 相较于对照中(siCTL), siRNA中PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平显著降低。图5 lncRNA CBR3-AS1激活HL-60细胞株的PI3K/AKT/mTOR/S6K通路
lncRNA CBR3-AS1位于羰基还原酶3(CBR3)的反义区域,在乳腺癌、胃癌、胆管癌等中均观察到lncRNA CBR3-AS1高表达[11-13]。然而, lncRNA CBR3-AS1在AML中的表达和功能报道较少。研究[14]表明, P13K/AKT/mTOR/S6K通路参与肿瘤细胞蛋白质的合成,在细胞的增殖、分化、代谢、生存等方面发挥重要作用。P13K/AKT/mTOR/S6K通路信号的持续激活导致细胞增殖信号过度活化和凋亡信号抑制,最终导致肿瘤细胞增殖失控。研究[15]表明,超过50%的AML中都存在PI3K/AKT/mTOR/S6K信号通路的异常活化。
在K562和HL-60细胞系中建立了一个耐药倍数接近或超过100倍的阿糖胞苷耐药模型,通过PCR定量的方法发现lncRNA CBR3-AS1在耐药细胞株中表达显著升高。在耐药株中利用siRNA敲低lncRNA CBR3-AS1可以极大降低阿糖胞苷的IC50, 逆转K562和HL-60的耐药表型。而在野生株中过表达K562和HL-60, 也可以极大提高阿糖胞苷的IC50, 诱导耐药表型。这些数据表明, lncRNA CBR3-AS1介导了阿糖胞苷在K562和HL-60中的耐药。蛋白印迹实验证实,过表达lncRNA CBR3-AS1显著提高了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平,敲低lncRNA CBR3-AS1显著降低了PI3K、AKT、mTOR、S6K等蛋白的磷酸化水平,提示lncRNA CBR3-AS1可能是通过激活P13K/AKT/mTOR/S6K通路诱导阿糖胞苷的耐药。
相较于其他长链非编码RNA, lncRNA CBR3-AS1在肿瘤耐药中的研究较少,且主要集中于其在肿瘤发生发展过程中的作用[16-17]。研究[11]表明, lncRNA CBR3-AS1在乳腺癌中通过调节c-Jun氨基末端激酶-1(JNK1)/丝裂原活化蛋白激酶(MEK4)介导的MAPK通路导致了阿霉素耐药,这提示lncRNA CBR3-AS1可能在包括阿霉素、阿糖胞苷在内的多种化疗药物耐药机制中具有相似的重要作用, lncRNA CBR3-AS1可能作为多重耐药机制的共同干预靶点。
本研究也存在不足, lncRNA CBR3-AS1可能通过激活PI3K/AKT/mTOR/S6K通路介导了在AML阿糖胞苷耐药的结论推导均基于离体的细胞学水平,在体研究中仍有必要进行验证。进一步的研究有必要从动物模型考察和分析lncRNA CBR3-AS1与PI3K/AKT/mTOR/S6K通路活性的关系。
综上所述, lncRNA CBR3-AS1可能通过激活PI3K/AKT/mTOR/S6K通路介导了在AML阿糖胞苷耐药,这为新药研发及临床治疗AML阿糖胞苷耐药提供了新的靶点和思路。
猜你喜欢
山东化工(2024年1期)2024-02-04 09:47:12
天津医科大学学报(2019年6期)2019-08-13 07:04:42
中外医疗(2016年15期)2016-12-01 04:25:34
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
天津护理(2015年4期)2015-11-10 06:12:02
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国当代医药(2015年16期)2015-03-01 02:03:11
中国医药导报(2015年27期)2015-02-28 22:08:02
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
遗传(2014年3期)2014-02-28 20:59:01
