
小儿咳喘灵颗粒质量标准优化
2022-05-16 07:03杨群肖文涛李韦
药品评价 2022年4期
杨群,肖文涛,李韦
九江市检验检测认证中心药品分中心,江西 九江 332000
小儿咳喘灵颗粒配方源自经典名方-麻杏石甘汤,是由张仲景所创,由七味药材组成,分别是金银花、麻黄、板蓝根、甘草、瓜蒌等,味甜、辛、微苦,能够清宣肺热、止咳祛痰[1],多用于治疗上呼吸道感染引发的咳嗽,能够有效改善支气管炎、肺炎、哮喘患儿的肺功能及临床症状[2-6],主要不良反应为呕吐及腹泻[7]。市售小儿咳喘灵颗粒的质量一致性相对较差[8],对其质量标准进行研究有利于提高质量标准和质量控制。目前小儿咳喘灵颗粒有批准文号100 多个,执行标准有部颁标准及不同企业注册标准,存在不同程度内容不完善、指标不合理、限度差异大等情况[8-11]。对小儿咳喘灵配方进行研究发现,方中麻黄为君药,能够宣肺、平喘、散寒[8,12]。麻黄碱是小儿咳喘灵制剂里面有明确药效的一种主要成分,量化控制较好。金银花味甘、性寒,能够消炎、清热、解毒,可治热毒血痢,感染性疾病,其活性成分为绿原酸[13-16]。甘草味甘、性平,能够补脾、祛痰、益气、止痛,可治脾胃虚弱,心悸气短[17-18]。瓜蒌在此前小儿咳喘灵颗粒的研究中未见报道。故本研究对瓜蒌、甘草进行薄层色谱鉴别研究,并采用高效液相色谱同时测定麻黄中盐酸麻黄碱和金银花中绿原酸含量,为进一步制定和完善小儿咳喘灵颗粒的质量标准提供数据支持。
1 仪器与试药
1.1 仪器
高效液相色谱仪[沃特世公司E2695,带紫外检测器(沃特世公司2489)];电子天平(赛多利斯公司BSA 124S、BP 211D);超声波清洗机(上海艺源超声设备有限公司YIY-UL300W-C);薄层色谱数码成像系统(瑞士卡玛公司TLC Visualizer)。
1.2 对照品
盐酸麻黄碱(批号:171241-201007,含量以99.7%计);绿原酸(批号:110753-201716,含量以98.0%计);甘草苷(批号:111610-201106);甘草对照药材(批号:120904-201620);瓜蒌对照药材(批号:121596-201903)。均购自中国食品药品检定研究院。
1.3 样品
小儿咳喘灵颗粒[葵花药业集团(襄阳)隆中有限公司,批号:191103,191204,191206]。
1.4 试剂
硅胶G(德国默克公司,批号:1056260001);薄层色谱板(德国默克公司);乙腈(色谱纯);水为超纯水;石油醚、乙酸乙酯、甲酸、冰醋酸、正丁醇均为分析纯。
2 方法与结果
2.1 薄层鉴别
2.1.1 瓜蒌的薄层鉴别分别取样品各2 g 于50 mL离心管中,加入40 mL 甲醇,混匀,超声处理20 min,收集滤液,蒸干后加20 mL 水复溶,提取复溶液时分4 次加入40 mL 正丁醇,收集正丁醇液、蒸干得残渣,再用2 mL 正丁醇溶解得供试品溶液。取瓜蒌对照药材,相同方法制成对照药材溶液。使用不含瓜蒌的模拟处方制成不含瓜蒌的空白样品,取空白样品,相同方法制成阴性样品对照溶液。吸取5 μL 供试品、对照药材、阴性样品对照溶液,分别点于同一硅胶G 薄层板上,展开剂为甲酸:乙酸乙酯:石油醚(60~90 ℃)(0.1∶1∶4),先展开条带,再取出,将展开后的薄层板晾干,均匀喷上10%的硫酸乙醇,加热显色至明晰(105 ℃),于365 nm 波长的紫外光灯下进行比对。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的主斑点,阴性无干扰。见图1。
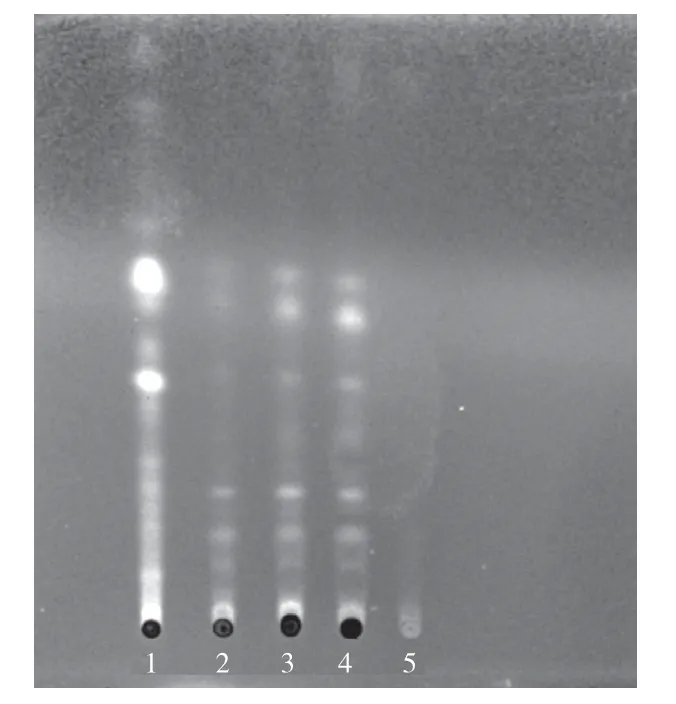
图1 瓜蒌鉴别图谱
2.1.2 甘草的薄层鉴别分别取样品各2 g于回流瓶中,加入40 mL 甲醇,混匀,加热回流30 min,滤过,收集滤液,蒸干后加20 mL 水复溶,提取复溶液时,分3 次加入60 mL 正丁醇,收集合并提取液,用20 mL水洗涤3 次,蒸干正丁醇液,得残渣,再用5 mL 甲醇溶解得供试品溶液。取0.5 g 甘草对照药材于回流瓶中,相同方法制得对照药材溶液。取甘草苷对照品,加甲醇溶解稀释得1 mg/mL 甘草苷对照品溶液。使用不含甘草的模拟处方制成不含甘草的空白样品,取空白样品,相同方法制成阴性样品对照液。吸取2 μL上述六种溶液,分别点于同一硅胶G 薄层板(1%氢氧化钠溶液制成)上,展开剂为乙酸乙酯∶水∶冰醋酸∶甲酸(15∶2∶1∶1),先展开条带,再取出,将展开后的薄层板晾干,均匀喷上10%的硫酸乙醇,加热显色至明晰(105 ℃),于365 nm 波长的紫外光灯下进行比对。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点,阴性无干扰。见图2。
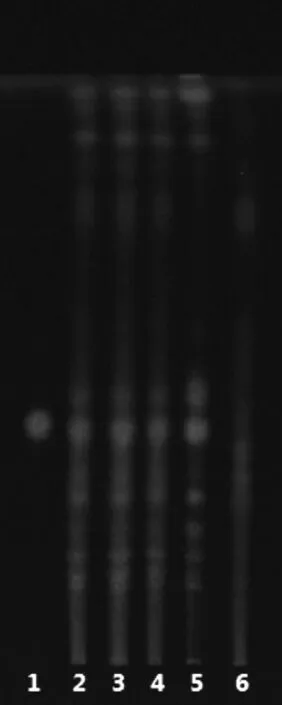
图2 甘草及甘草苷鉴别图谱
2.2 含量测定
2.2.1 高效液相法色谱条件色谱柱:Venusil MP C18柱(4.6 mm×250 mm,5 μm);流动相:乙腈-0.1%磷酸,梯度洗脱,见表1;流速:1.0 mL/min;检测波长:0~15 min,207 nm;15~25 min,327 nm;25~40 min,207 nm;柱温:25 ℃;进样量:10 μL。

表1 流动相梯度洗脱程序表
2.2.2 对照品制备精密称取15.79 mg 盐酸麻黄碱对照品,置25 mL 的容量瓶中,超声15 min,加甲醇定容,得629.7 μg/mL 的盐酸麻黄碱储备液。精密称取绿原酸对照品9.32 mg,置25 mL 的容量瓶中,超声15 min,加甲醇定容,得365.3 μg/mL 的绿原酸储备液。
2.2.3 样品制备取样品研细,精密称取2 g,加入甲醇25 mL,称重,超声20 min(功率300 W,频率25 kHz),放冷后再次称重,用甲醇补足减少的重量,先摇匀,再滤过,然后取续滤液,即得样品溶液。
按照颗粒处方比例和制备工艺制备出缺麻黄和金银花的阴性对照制剂,同样使用“样品制备”的方法制得阴性对照溶液。
2.2.4 线性关系考察取绿原酸对照品储备液,加入适量甲醇稀释,制得1mL 中含18.3、36.6、73.2、183.0、366.0 μg 绿原酸的标准系列溶液,使用高效液相色谱仪进行测定,以其质量浓度作为横坐标,峰面积作为纵坐标,得出标准曲线。结果:回归方程Y=21 084.595 2X+28.329 1,相关系数为0.999,说明绿原酸在18.3~366.0 μg/mL 范围内符合线性要求。
取盐酸麻黄碱对照品储备液,加入适量甲醇稀释,制得1 mL 中含6.3、12.6、25.2、63.0、126.0 μg盐酸麻黄碱的标准系列溶液,使用高效液相色谱仪进行测定,以其质量浓度作为横坐标,峰面积作为纵坐标,得出标准曲线。结果:回归方程Y=25 351.509X-5 999.754 3,相关系数为0.999,说明盐酸麻黄碱在6.3~126 μg/mL 范围内线性条件良好。
2.2.5 精密度考察取浓度为182.7 μg/mL 的绿原酸对照品溶液,按上述色谱条件连续测定6 次峰面积,相对标准偏差(RSD)为0.38%。取浓度为62.97 μg/mL 的盐酸麻黄碱对照品溶液,按上述色谱条件连续测定6 次峰面积,RSD 为0.38%,符合精密度要求。
2.2.6 稳定性考察取样品2 g,精密称定,按“2.2.3”的方法制备样品溶液,分别放置0,2,4,8,12,24 h 后进样测定,绿原酸峰面积RSD 为1.2%,盐酸麻黄碱峰面积RSD 为1.1%,24 h 内稳定性良好。
2.2.7 加样回收率实验取六份样品,每份2.0 g,精密称定,分别置25 mL 量瓶中,各加入含盐酸麻黄碱对照品(0.113 mg/mL)、绿原酸对照品(3.024 mg/mL)0.8、0.8、1.0、1.0、1.2、1.2 mL,加入 甲醇15 mL,超声(功率300 W,频率25 kHz)处理20 min,先定容,再摇匀,然后滤过,按色谱条件来测定含量,计算回收率。见表2,表3。
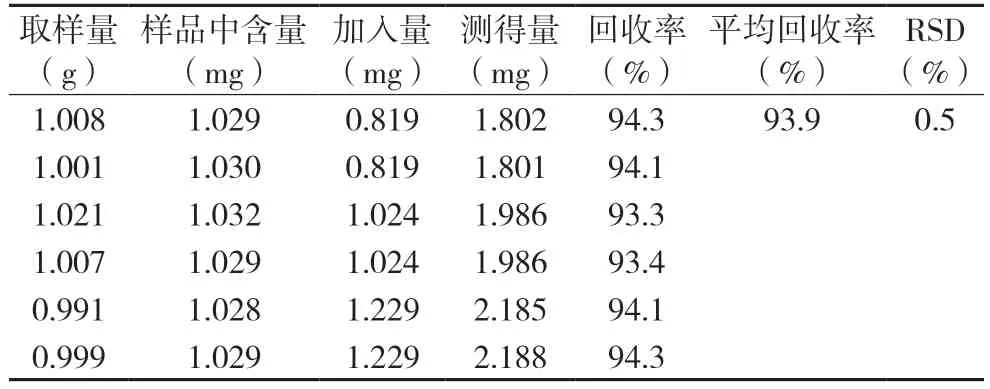
表2 绿原酸回收率
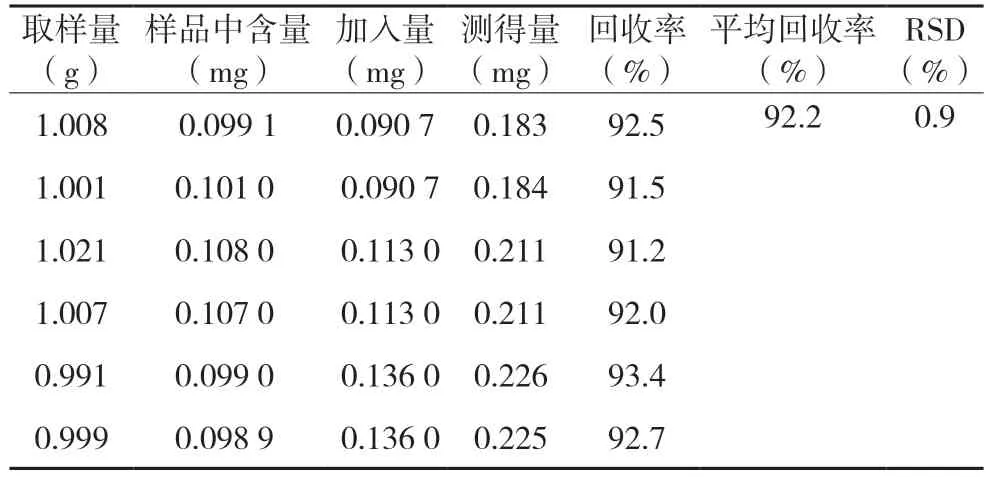
表3 盐酸麻黄碱回收率
2.2.8 检出限测定经测定盐酸麻黄碱的检出限0.006 3 μg/mL,绿原酸的检出限为0.000 36 μg/mL。
2.2.9 样品含量测定精密称取适量小儿咳喘灵颗粒样品,按“2.2.3”的方法制备样品溶液,按“2.2.1”项下色谱条件重复进样测定4 次,计算样品中2 种成分的含量,结果见表4。
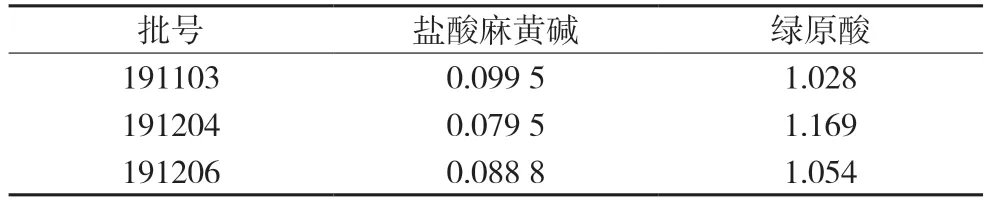
表4 样品测定含量(mg/g)
3 讨论
查阅文献,小儿咳喘灵颗粒中有检测麻黄、金银花等[19-23]的相关报道,但尚未见对瓜蒌有任何研究,对样品质量控制存在空白。本研究对瓜蒌进行TLC 鉴别研究,曾选用乙酸乙酯:甲酸:甲醇:水(12∶0.1∶1∶0.1)作为展开剂,但效果不好,有阴性样品干扰,易造成假阴性。通过反复多次试验,最终确定使用甲酸:乙酸乙酯:石油醚(60~90 ℃)(0.1∶1∶4)作为展开剂,展开效果良好,阴性无干扰。通过对18 家企业18 批样品检验,结果有2 家样品未检出与瓜蒌对照药材相同颜色的斑点,由于目前尚未见有任何标准对此有规定,提示是否存在投料风险。
对于甘草鉴别,参考文献,比较了甲醇超声和甲醇回流后再用饱和正丁醇提取两种方法,前种方法阴性样品有一定的干扰,后种方法特异性强,阴性无干扰。
测定木犀草苷、绿原酸大多使用高效液相色谱法[15,22],本研究将绿原酸和盐酸麻黄碱两个成分在一个系统中检测出来,曾选用甲醇-0.1%磷酸、乙腈-0.1%磷酸和乙腈-水溶液(15∶85,用磷酸调节至pH3.0),最后确定的流动相为乙腈-0.1%磷酸溶液,用梯度洗脱的方式,取得较好分离效果,做到了检测效率的提高和试验成本的降低。
通过多批次样品的调研检验发现,部颁标准执行覆盖面最广,但项目设置不完善,仅有两个理化鉴别项目,无法有效控制药品质量。各注册标准项目设置差异大,项目设置不完善,相同指标方法及规定限度差别较大。为进一步完善统一现行质量标准,本研究增加制剂中瓜蒌、甘草的薄层鉴别,增加麻黄、金银花的有效成分指标控制,望能帮助企业提高产品质量。
对18 家企业50 批样品进行检验,其中绿原酸、麻黄碱最高含量分别为5.056、0.519 mg/g,最低含量分别为0.114、0.041 mg/g,分别相差了44 倍和12 倍。不同厂家乃至同一厂家不同批次生产的样品中各成分含有量均有较大差异,质量参差不齐,可能与药材产地、采收季节、生产工艺等因素有关,具体原因有待进一步研究,提示相关监管研究部门引起重视。
猜你喜欢
当代水产(2021年9期)2021-12-02
中老年保健(2021年9期)2021-08-24
家庭百事通·健康一点通(2020年8期)2020-09-08
广西植物(2020年5期)2020-07-04
新传奇(2019年19期)2019-10-08
新传奇(2019年16期)2019-10-08
新传奇(2019年17期)2019-08-04
中国民族民间医药·下半月(2014年2期)2014-09-26
中国民族民间医药·下半月(2014年4期)2014-09-26
