
碱性电催化析氢催化剂的研究进展
2022-05-16 09:25武美霞陈龙刚李作鹏
山西大同大学学报(自然科学版) 2022年2期
武美霞,陈龙刚,李作鹏,郭 永
(山西大同大学化学与化工学院,山西大同 037009)
可再生清洁能源的研发受到了社会和科学界的广泛关注。目前,科学界公认的生产清洁能源尤其是绿氢的的方式主要是电催化和光电催化。可再生清洁能源——从简单的氢气到复杂的能量载体(如C2 分子和氨)都可以在温和条件下通过电化学或光化学过程从水、二氧化碳和氮气中获取[1]。其中,碱性析氢反应(hydrogen evolution reaction,HER)作为碱性条件下电解水的半电池反应具有举足轻重的作用。其反应机理包括水解离(Volmer)与质子还原(Heyrovsky 或Tafel)两个步骤,是研究与水分子相关还原反应中普遍规律的关键所在[2]。深入探究碱性析氢反应的机理,可为后续研究复杂产物的合成奠定坚实的基础。
近年来,科学家们虽然对酸性HER 进行了深入的机理剖析,但对碱性HER 的反应机理依然困惑不已。有研究表明,碱性HER速度比酸性环境慢2~3个数量级[3]。人们普遍认为,除了碱性HER 中的氢中间产物(H*,*代表吸附位点)是通过水离解步骤生成外,酸性HER 和碱性HER 的反应机理相似。因此,碱性HER 的反应机理也包含两个步骤:(i)H2O+e-→H*+OH-(Volmer);(ii)H2O+e-+H*→H2+OH-(Hey⁃rovsky)或2H*→H2(Tafel)。尽管科学界已经提出了一些理论,但pH 影响HER 活性的机理,目前依旧不明确。Markovic 等人提出在Pt 表面水解离引入的附加能垒决定了碱性HER的速率[3],而Sheng等人指出,催化剂和H*中间体之间的结合能决定了反应动力学[4]。还有一些其他影响因素的文献报道,如零自由电荷电位(pzfc)和通过电化学双层区域的阴离子转移效率。这些理论都集中于回答有关碱性HER 反应机理的两个关键问题:①缓慢动力学的真正原因是什么;②与其他因素(如HBE 和pzfc)相比,水解离过程在多大程度上影响整体反应动力学。
由于反应机理尚不清楚,碱性HER 催化剂的开发仍主要依靠反复试验积累经验。特别是在不确定反应所涉及的中间体(即H*、OH*和H2O*)的影响权重的情况下,设计催化剂时就需要考虑所有中间体的吸附情况。人们希望通过调控材料的电子和物理结构设计具有最低活化势垒和与中间产物相互作用力适宜的催化剂。密度泛函理论(DFT)等计算方法通常用于阐明不同材料的固有特性与HER 性能之间的定量关系(如H 吸附能力-活火山图)。尽管这些结果似乎与酸性HER 数据完全吻合,但在碱性环境下对其进行预测时,这些结果并不准确。这是因为,在碱性电解液中,HER 反应活性很可能不仅仅由一个单一的速率控制步骤确定。另外,和碱性与酸性环境相比,其他因素(如反应界面、电化学环境和催化剂的物理杂化结构)对反应过程的影响更大[5]。本文简要介绍在碱性条件下HER 机理的最新发展,讨论如何通过调控电子结构或构建活性位点,设计高活性碱性HER催化剂。
1 碱性条件下HER动力学缓慢的缘由
Pt 基材料是酸性条件下HER 最有效的催化剂,然而在碱性条件下Pt 催化活性同比却下降2~3 个数量级。目前有三种理论解释其原因,即水解离理论、氢结合能理论(HBE)和零电荷电位理论。
水解离理论认为OH*的吸附与H*吸附存在竞争,导致H2析出能力变弱。于是设计通过提供OH*吸附位点来促进H*的产生,从而促进Volmer 步骤。双活性位点催化剂增加了一种组分,可能引起催化剂表面结构和电子结构的变化,从而影响HER 的活性。另一方面,OH*的吸附不参与Volmer 步骤,所以也不影响碱性析氢活性,但是OH-的作用可能与碱性条件下HER 动力学缓慢相关。不同于水解离理论强调OH*的吸附作用,Jia 等认为OH*扩散到双电层外才是反应决速步骤[3]。通过改变电解质中的阳离子(如以锂离子替代钾离子),能够促进OHad转移,从而提高整体碱性析氢效率。
氢结合能理论认为碱性HER 与酸性HER 一样,其反应活性与催化剂-H 之间的相互作用密切相关。但是HBE 理论的适用范围并不广泛,例如Pt(111)晶面在不同pH下析氢活性明显改变,但是其Hupd位置基本不变,它也不仅仅与H 吸附相关。虽然理论计算证明吸附的OH*、H2O*等活性物种与催化剂的HBE相关,但还缺乏相应的实验证据。
零电荷电位(pzfc)理论认为HER 反应速率取决于电极电势和pzfc 的关系。Hupd 与pzfc 同向改变,催化剂表面水的排列也随之发生改变,从而控制H+或者OH-由催化剂表面到电解质溶液的迁移。通过将过渡金属Ni 引入Pt(111)晶面,pzfc 可以更接近H吸附区域的电位,从而减少了H 吸附的活化能,促进整体的HER 速率。该结果从催化剂-H 相互作用角度,为所谓的“双活性位点”催化机理提供了合理的解释。
2 碱性HER电催化剂的设计
2.1 构建双活性位点
虽然OH-在碱性HER 中的特定作用尚不清楚,但可以肯定的是,催化剂与OH-之间的相互作用是影响催化活性的关键因素之一(图1)。然而,由于这两种中间体的结合能之间的关系尚未可知,因此在一个位点上平衡OH*和H*的吸附/解吸能力是极其困难的。更切实际的策略之一就是设计构建两个活性位点来分别体现其独特的功能。

图1 碱性条件下可能的反应机理示意图
根据这一理念,Markovic 等人报告了一系列催化剂,通过用另一种金属或金属氢氧化物位点修饰本体Pt[6]。总的想法是通过将酸性HER 火山区顶部的材料(如Pt)与能够为水解离过程提供良好OH-相互作用位点的亲氧金属耦合,实现具有适当HBE 的混合材料。与HBE 的情况一样,催化剂的OH-结合能必须既不太强也不太弱,以提供适当的催化剂-水相互作用。因此,镍、钌和钴等因其适当的亲氧性脱颖而出,被广泛应用于双活性位点催化剂的设计中,并表现优异的催化性能。基于双中心合金的成功设计,非贵金属材料已被用于取代Pt 作为H*相互作用中心,以构建更具成本效益的催化剂。Yang 等人使用MoS2作为H 活性材料,使用层状双氢氧化物(LDH)作为OH 活性位点,以获得良好性能(图2)[7]。DFT 计算表明,MoS2/LDH 异质结构性能的提高是由于活化能降低,这是优化水离解过程的结果。与Pt/Ni(OH)2一样,MoS2/LDH 为H 吸附(在MoS2上)和水离解(在LDH 上)提供了不同的位点,这些位点协同促进整个碱性HER 过程。与H 吸附位点类似,OH 相互作用位点也可以设计或用几种材料替代。基于适当平衡水离解和氢吸附的思想,成功设计具有高碱性HER 活性的类似双中心催化剂,被证明是提高碱性HER催化剂活性的有效策略。

图2 碱性条件下MoS2/LDH界面HER的示意图
2.2 调控催化材料电子结构
对于大多数多相催化剂,电子结构是决定其对各种中间体吸附能力的唯一因素。因此,调控催化剂的电子能带是改善其催化性能的有效方法。特别是对于碱性HER,调整催化剂的电子结构主要是为了改善其HBE。Huang 等人报道了一种PtNi/NiS 纳米线,其在-0.07 V 电压下的电流密度是Pt/C 的5.58倍[8]。Zheng等人报道了一种六边形紧密堆积纳米多极体形式的PtNi合金,显示其活性比Pt/C好得多(图3)[9]。
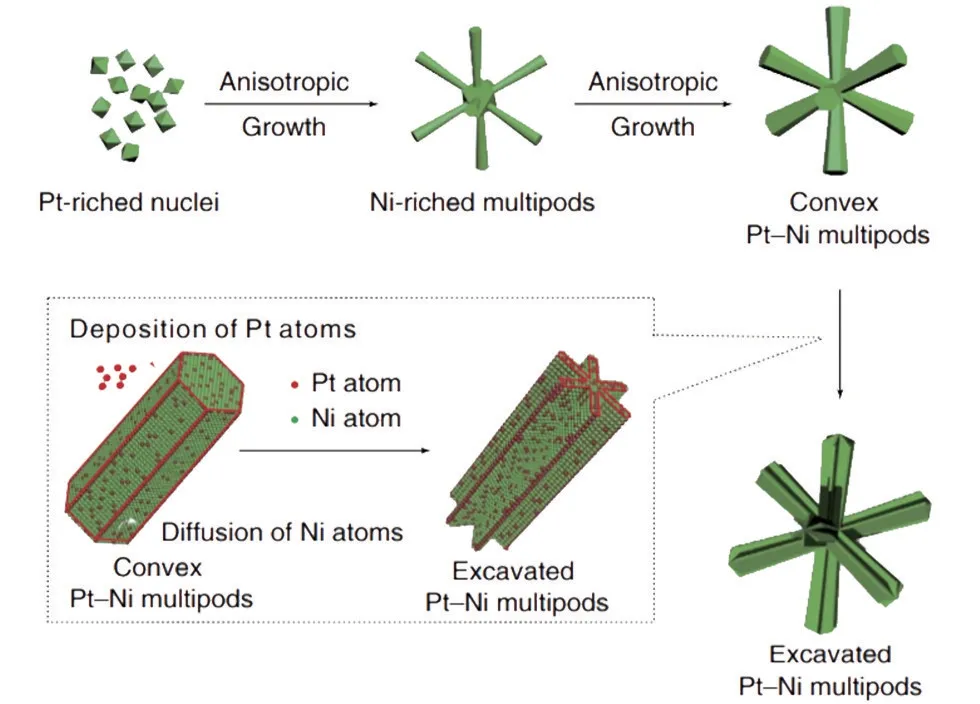
图3 Pt-Ni纳米多极体形成的示意图
对大多数金属催化剂,通过合金化另一种金属,HBE就能被有效调控。对于HER 活性较差的两种金属材料,合金化以后有可能达到优异的性能。Cu-Ti双金属电催化剂表现出卓越的碱性HER 活性[10]。通过调整催化剂中Ti的浓度,改变了CuTi的电子结构,并将催化剂的HBE 优化到更合适的水平,从而将该催化剂置于火山图的顶部。因此尽管Cu或Ti都不是很好的HER 候选者,但Cu-Ti 催化剂表现出超强的活性。MoNi4合金也出现了类似情况。尽管Mo、Ni和MoO2表现出非常缓慢的碱性HER 动力学,但由于合金的电子结构重建,MoNi4合金的电子结构显著改善,并表现出出色的水离解能力。另一种提高过渡金属吸附能力的常用策略是改变催化表面的电荷密度。实现这一目标最常用的方法是将过渡金属基催化剂与其他材料复合,比如缺陷调控和原子掺杂等。还有报道构建Mo2C 和N 掺杂C 复合材料,调控电子从Mo2C转移到N上协同促进碱性HER催化性能[11]。
2.3 调节催化剂表面几何结构
碱性HER 对催化剂的表面结构极其敏感。早在20 世纪90 年代,Markovic 等人就报道了Pt 不同单晶面上的HER/HOR 非常不同,在碱性环境中催化活性活性顺序为Pt(111) <Pt(100) <Pt(110)[12]。尽管DFT计算阐明了不同的Pt 晶面如何改变催化剂的电子结构和H 吸附能力,但碱性HER 的真正表面敏感性仍然是一个谜。到目前为止,催化剂表面几何结构如何分别影响Hupd和Hopd还不清楚。实际上,晶格应力是调控催化剂表面几何结构最有效的方法之一。通常通过晶格调整构建晶格缺陷等改变各种催化材料的表面几何结构。这种方法已被用于提高ORR 催化剂的催化性能,并被证明在酸性环境中对MoS2和WS2等HER 催化剂是有效的。近年来,许多人试图将这种方法推广到碱性HER应用中。Pt3Ni合金是通过构建Pt3Ni 多面体,然后去除多余的镍,并在Ar 气氛中退火,构建了Pt3Ni 纳米骨架结构[13]。通过Ni 对Pt 的应力调控(Ni 在Pt 内部),优化了Pt 的电子结构,表现出优异的碱性HER 催化活性。另外,将应力调控的核壳双金属Ru@Pt材料与应力可忽略的RuPt合金相对比,再次证明应力调控可以增强催化剂-OH相互作用并减弱催化剂-H 吸附能力,从而提高碱性HER 催化活性[14]。除了对金属表面几何结构的调控以外,应力控制也可以用于非金属纳米材料中,如Ling 等报道的S-CoO 富含锯齿的纳米棒[15],通过应力调控使得S-CoO 纳米棒产生了大量的O-空位,优化了结构,提升了碱性HER 活性,甚至与商业金属催化剂相当。
3 结语
由于理论计算和材料工程技术的发展,已经有大量的高活性碱性HER 催化剂不段涌现,但仍然面临以下几个挑战:①碱性HER 的缓慢动力学的本质原因仍然未知;②在碱性HER 过程中,离子通过双电层/水层的转移被证明是至关重要的,但目前还没有相关的有针对性的优化设计;③纳米结构催化剂碱性HER 的反应机理研究不足。为了解决这些问题,在后续研究中先进的方法如原位表征和理论计算等应予以重视,以监测和模拟真实反应过程,有助于理解碱性HER 机理,从而更好地设计高活性和稳定性的先进碱性HER催化剂。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
家庭科学·新健康(2022年7期)2022-07-13
汽车实用技术(2022年5期)2022-04-02
表面技术(2022年1期)2022-02-12
当代陕西(2020年23期)2021-01-07
电脑知识与技术(2018年19期)2018-11-01
智富时代(2018年8期)2018-09-28
智富时代(2018年8期)2018-09-28
