
儿童髓鞘少突胶质细胞糖蛋白抗体相关急性播散性脑脊髓炎的临床特点
2022-05-11 05:36:24吴小慧庄嘉鑫林学锋
海南医学 2022年8期
吴小慧,庄嘉鑫,林学锋
泉州市儿童医院小儿神经内科,福建 泉州 362000
髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)表达于中枢神经系统少突胶质细胞细胞膜上,位于髓鞘的最外层,含量极微,具有免疫原性,参与髓鞘的完整性、黏附、细胞表面交互作用等[1]。MOG 抗体介导的炎性脱髓鞘疾病是一类不同于多发性硬化(multiple sclerosis,MS)、AQP4阳性的视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)的独立疾病谱,命名为抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G抗体(anti-myelin oligodendrocyte glycoprotein-IgG,MOG-IgG)相关疾病(MOG-IgG associated disorders,MOGAD)[2]。MOGAD
可表现为多种不同的临床表型,包括视神经炎(optic neuritis,ON)、脑膜脑炎、脑干脑炎、脊髓炎、急性播散性脑脊髓炎(acute disseminated encephalomyelitis,ADEM)等,儿童及成人均可发病,并表现为年龄相关性特征,儿童多表现为ADEM,成人多表现为ON、脊髓炎和脑干脑炎[3-6]。随着MOG抗体检测的开展,MOGAD报道越来越多,也越来越受到临床医生的关注。本研究对儿童MOG抗体相关ADEM的临床特点进行总结分析,以进一步提高临床医师对本病的认识。
1 资料与方法
1.1 一般资料 回顾性分析2019 年5 月至2020年12月在泉州市儿童医院小儿神经内科住院的18例MOG 抗体相关ADEM 患儿的临床资料,包括患儿的基本资料、临床表现、实验室检查、头颅及脊髓核磁共振(magnetic resonance imaging,MRI)检查结果、治疗疗效及预后随访情况。所有入选患儿均排除感染、肿瘤、遗传代谢病、线粒体病等疾病。本研究已通过医院医学伦理委员会批准。
1.2 诊断标准 符合《IPMSSG 儿童多发性硬化和免疫介导的中枢神经系统脱髓鞘疾病诊断标准(修订版)》(2012)[7]中关于ADEM 的诊断标准,并参照文献[2,8]关于MOGAD的诊断标准。
1.3 MOG抗体的检测及其他辅助检查 所有患儿均在免疫治疗前的急性发作期应用德国欧蒙公司的间接免疫荧光转染细胞(CBA)法检测血清及脑脊液MOG-IgG 抗体,同时检测血清及脑脊液AQP4 抗体、抗N-methyl-D-aspartate receptor (NMDAR)抗体。所有患儿均行脑脊液常规、生化、寡克隆区带及病原学检测,均完成头颅/脊髓MRI检查。其他辅助检查包括血乳酸、血氨、抗核抗体谱、甲状腺抗体、脑电图。
1.4 治疗方法 所有患儿急性期均予静脉丙种球蛋白(intravenous immunoglobulin,IVIG) (2 g/kg,总量分2~5 d 静脉滴注)、静脉滴注甲泼尼龙(intravenous methylprednisolone,IVMP) [20 mg/(kg·d),最大量1 g/d]冲击治疗3 d,间歇期序贯口服泼尼松[1.5~2 mg/(kg·d),最大量60 mg/d]4 d,7 d 为一个疗程,共3个疗程。缓解期继续口服泼尼松[1.5~2 mg/(kg·d)],最大量60 mg/d),每个月予IVIG(1 g/kg,分1~2 d 静脉滴注),泼尼松足量6~8 周后根据病情缓慢减量,并以小剂量维持。复发患儿予复查血清MOG-IgG 抗体及头颅MRI,并再次予IVIG 及IVMP 冲击治疗,以后泼尼松酌情减量并小剂量维持,配合每个月予IVIG(1 g/kg,分1~2 d 静脉滴注)。所有患儿均未使用免疫抑制剂,均未使用抗癫痫药及抗生素。
1.5 随访方式 所有患儿出院后均于小儿神经内科专科门诊随访。出院1 个月后随访1 次,此后每3个月随访1次,进行神经系统专科查体,根据病情复查血清MOG-IgG抗体及影像学检查。
2 结果
2.1 一般情况 18例患儿中男性10例,女性8例;发病年龄2.5~8 岁,中位年龄4.6 岁,5 岁以下12 例(66.7%);发病前1个月内有前驱感染史10例(55.6%),发病前3个月内有疫苗接种史3例(16.7%)。所有患儿既往均健康,无特殊服药史,无中毒史,均无特殊家族史。
2.2 临床表现特点 所有患儿均急性起病,均有脑病症状(表现为不同程度的意识障碍、精神行为异常)(100%),其他包括抽搐8 例(44.4%,8/18),发热(体温>37.5℃,持续1~15 d)7 例(38.9%,7/18),共济失调7 例(38.9%,7/18),头痛呕吐6 例(33.3%,6/18),震颤3 例(16.7%,3/18),肢体瘫痪2 例(11.1%,2/18),构音障碍1例(5.6%,1/18),所有患儿均无视力障碍、括约肌功能障碍及颅神经损害表现。
2.3 实验室检查 血清MOG-IgG 抗体滴度(1∶10)~(1∶320),脑脊液MOG抗体滴度阴性~1∶10。脑脊液细胞数升高16 例(88.9%),其中细胞数(0.022~0.195)×109/L[正常(0~0.010)×109/L],淋巴细胞升高为主,蛋白轻度升高3 例(16.7%),蛋白475~550 mg/L(正常150~450 mg/L),糖、氯化物均在正常范围,脑脊液寡克隆区带阳性仅1 例(5.6%),脑脊液NMDAR抗体均阴性,脑脊液病原学检查均阴性。所有患儿血清及脑脊液AQP4抗体均阴性。11例患儿行抗核抗体谱检查,结果均阴性。10例患儿行甲状腺功能及甲状腺抗体检查,甲状腺抗体均阴性,2例游离三碘甲状腺原氨酸(FT3)降低,1例游离四碘甲状腺原氨酸(FT4)降低,均未经特殊处理,出院前复查均降至正常。13 例患儿行脑电图检查,10 例表现为背景活动弥漫性慢化,其余3例正常。
2.4 影像学检查 所有患儿头颅MRI均异常,表现为多发斑片状异常信号影,T1WI 呈等信号或低信号,T2WI、T2Flair呈高信号,DWI呈等信号或高信号,直径多大于2 cm,边缘模糊,部分可伴强化。急性期共行20例次头颅MRI检查,病变受累最多见为皮质下白质(70.0%,14/20),其余为深部脑白质(55.0%,11/20)、皮质(45.0%,9/20)、丘脑(40.0%,8/20)、基底节(40.0%,8/20)、脑干(30.0%,6/20)、脑室旁白质(25.0%,5/20)、第四脑室周围(25.0%,5/20)、小脑(20.0%,4/20)、胼胝体(10.0%,2/20)、半卵圆中心(10.0%,2/20)、内囊(5.0%,1/20)、小脑脚(5.0%,1/20)。其中1 例表现为双侧额顶颞枕叶、双侧半卵圆中心、左侧丘脑及胼胝体压部左侧多发斑片状异常信号,以双侧顶枕叶为主,呈大片状改变,类似“脑白质营养不良”样改变(图1 G~1I)。脊髓受累1 例,为颈段至胸段长节段病变。患儿的影像学检查结果见图1。
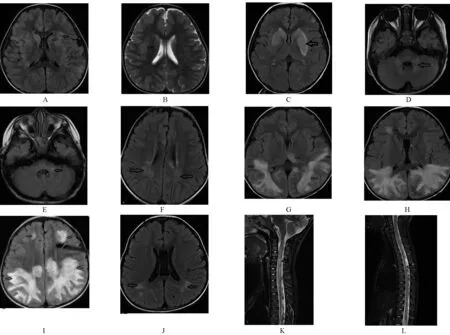
图1 MOG抗体相关ADEM患儿的MRI检查结果
2.5 治疗结果及随访情况 所有患儿急性期予IVIG(2 g/kg)、IVMP(20 mg/kg)治疗后病情均改善,缓解期继续口服泼尼松,并以小剂量泼尼松维持。所有患儿起病后1个月复查头颅MRI病灶均吸收。2例未配合每月IVIG的患儿出现复发,复发间隔分别为首次发病后2个月、6个月,首次发病急性发作期血清MOG抗体均为1:320,分别于激素减量至30 mg/d、15 mg/d时复发,均为ADEM样表现,复查血清MOG抗体仍为1:320,复查头颅MRI 出现新发病灶,再次予IVMP 冲击及IVIG 治疗,病情均改善,此后小剂量泼尼松维持并配合每月IVIG(1g/kg),分别随访1 年10 个月、1 年均未再出现复发,复查头颅MRI 病灶均吸收。所有患儿随访过程中均未符合MS 的诊断。目前随访时间3个月~1年10个月,仅1例遗留轻微步态不稳,所有患儿均未遗留癫痫、肢体瘫痪、视力障碍、认知功能障碍、行为问题等。
3 讨论
MOGAD的发病机制尚未明确,细胞免疫和体液免疫机制可能均参与发病,并且有补体机制的参与[9]。研究发现,儿童期发病的汉族MOGAD病例可能与人类白细胞抗原(human leucocyte antigen,HLA) DQB1*05:02-DRB1*16:02 有关[10],并且其病情更易复发,预后更差。动物研究发现,注射人源MOG 抗体可产生实验性自身免疫性脑脊髓炎,由T细胞介导,产生超急性炎症反应,形成广泛的脱髓鞘斑块,并影响轴突蛋白的表达,并未增加轴突缺失、神经元或星形胶质细胞的死亡[11]。病理学研究发现,MOGAD 具有独特的免疫病理改变[12]。因此,MOGAD具有不同于MS、AQP4阳性的NMOSD的发病机制。MOGAD的临床表型在儿童存在双峰分布的特点,8 岁以下主要表现为ADEM,8 岁以上多表现为ON,且5 岁以下ADEM 患儿MOG抗体检出率最高[3-4,13-14]。
本研究中MOG抗体相关ADEM患儿的中位发病年龄4.6岁,5岁以下占66.7%,男女比例接近1:1,与多数文献报道一致[5,15-17]。文献报道MOG 抗体相关ADEM以5岁以下儿童尤为多见[3,14-16]。李小晶等[17]研究表明,MOG 抗体相关的ADEM 起病年龄5 岁左右。本研究亦体现了这一年龄特征。本研究中所有患儿均具有不同程度的意识障碍、精神行为异常等脑病症状,其他症状包括抽搐、头痛、小脑共济失调、肢体瘫痪、震颤等。其中抽搐症状在本研究中占44.4%,是除脑病外较常见的神经系统症状。研究发现,相比与NMOSD,MOGAD 的临床表现更常见发生癫痫发作[18]。癫痫发作可发生于疾病的任何阶段,甚至可早于其他症状出现[19]。因此对于某些不能用其他病因解释的癫痫发作,应考虑到MOGAD 的可能,及时行血清MOG-IgG的检测。
MOGAD 最初被认为呈单相性病程,随着研究的深入及随访时间的延长,多数研究发现MOGAD存在复发倾向[2-3,5,15-16],复发率报道不一(20%~88%)[2-3,5,15-16],复发与抗体滴度及抗体持续时间有关[2,16,20-22]。复发间隔时间1个月~4年不等,多数在起病1年内,平均复发间隔约5 个月[3,14,16,22]。LÓPEZ-CHIRIBOGA 等[2]研究发现,MOG 抗体持续阳性的ADEM 儿童复发率达88%,在6年的随访中发生22次复发。本研究中MOG抗体相关ADEM 2例出现复发,复发率为11.1%,复发率低于多数文献报道,复发病例的MOG 抗体滴度较高(1:320)。复发间隔时间分别为2个月、6个月,其中1 例符合多相性播散性脑脊髓炎(multiphasic disseminat edencephalomyelitis,MDEM)的诊断[7]。WATERS等[23]研究发现,MOG 抗体相关疾病中,ADEM 复发率较ON 及脊髓炎低,复发多发生于抗体持续阳性的病例,但少数亦可发生于抗体滴度不高或随访过程中抗体已转阴的病例,而另一方面,抗体持续存在亦可不复发。因此,MOGAD 复发的影响因素还需更多样本的研究及更长时间的随访观察。
MOG抗体相关ADEM的头颅MRI表现为颅内多发斑片状异常信号影,T2WI、T2FLAIR 高信号,范围较大,边界不清,一般呈非对称性,幕上和幕下均可累及,幕上受累部位包括脑室周围白质、深部脑白质、丘脑、基底节、胼胝体、皮层下白质等,可有软脑膜强化;幕下受累部位包括大脑脚、脑桥、脑桥被盖、延髓、小脑半球、小脑脚等[3,14]。脊髓病变可表现为长节段脊髓炎或非长节段脊髓炎,受累部位以颈髓最多见,其次为胸髓,腰骶椎和圆锥亦可累及[3,14]。本研究中患儿的头颅MRI改变幕上病变以皮质下白质受累最常见,幕下病变以脑干受累最常见。其中1 例病灶呈大片状,表现为类似“脑白质营养不良”样改变。HACOHEN等[24]亦报道了头颅MRI 表现为“脑白质营养不良”样改变的病例,需注意与遗传性脑白质病、线粒体病、某些代谢性疾病鉴别。本研究中有1 例同时脊髓受累,表现为长节段脊髓炎,包括颈段及胸段。
本研究中,脑脊液检查无特异性,细胞数可正常或升高,以淋巴细胞为主,蛋白正常或轻度升高,糖和氯化物均正常,仅1 例(5.6%)脑脊液寡克隆区带呈阳性。文献报道,MOGAD 患儿的脑脊液寡克隆区带阳性率不高[5,15,25],FERNANDEZ-CARBONELL等[14]研究发现,MOG抗体阳性的病例,其寡克隆区带阳性率与MOG抗体阴性的对照组相比并无差别。
MOGAD 急性期的一线治疗方案包括大剂量IVMP 冲击治疗、IVIG、血浆置换等,治疗反应均较好。缓解期治疗包括小剂量激素维持、免疫抑制剂、每月IVIG等[3,6,15,25-26]。本研究中所有患儿急性期均使用大剂量IVMP、IVIG 治疗,病情均得到缓解。缓解期治疗主要是小剂量激素维持,配合每月IVIG,均未使用免疫抑制剂。其中2 例出现复发,在每月使用IVIG 并予小剂量激素维持治疗后未再出现复发。其余16 例急性期后均规律每月使用IVIG,均未出现复发。HACOHEN 等[25]研究表明,IVIG 可减少MOGAD复发率,并改善预后。IVIG是唯一不会产生免疫抑制的治疗方法。一项体外试验研究发现,在MOG抗体介导的动物模型上,丙种球蛋白可保护髓鞘,预防髓鞘少突胶质细胞的死亡,并以剂量依赖的方式起作用[27]。而RAMANATHAN 等[15]研究发现,充分使用IVIG 亦可出现复发,复发可发生于减少IVIG剂量或延长使用间隔时间后。因此IVIG 是否能预防复发以及使用剂量及间隔时间仍需更大样本及更多的临床对照研究证实。研究发现,MOGAD对激素依赖,常在激素减量过程中或停药后复发,缓慢减量、小剂量维持可减少复发[5,15,21,28]。RAMANATHAN 等[15]研究发现,相比与使用其他免疫抑制剂,小剂量激素维持治疗或每月IVIG 治疗较少出现复发。但小剂量激素维持治疗的疗程、每月IVIG治疗的疗程目前尚无统一的方案。本研究中,MOG抗体相关ADEM复发率较低,并且复发病例未使用免疫抑制剂,使用小剂量激素维持配合每月IVIG 后未再出现病情反复。因此小剂量激素维持配合每月IVIG可能是减少复发的有效方案,但仍需更大样本及更多的临床对照研究证实。
MOGAD 的预后总体较好[15,16-17,23,29],预后可能与急性期的病程轻重及病灶受累范围有关[15-16]。ARMANGUE等[16研究发现,MOG抗体阳性的ADEM相比非ADEM 脑炎预后较好。HINO-FUKUYO 等[30]对未使用免疫抑制剂的儿童MOGAD 的长期随访观察发现,预后均较好,未遗留神经系统后遗症。本研究中,MOG抗体相关ADEM均未使用免疫抑制,预后均较好,仅个别遗留轻微步态不稳,均未遗留癫痫、肢体瘫痪、视力障碍等。POLAT İ等[21]研究发现,头颅MRI表现为“脑白质营养不良”样改变的MOG 抗体相关ADEM患儿,其预后较差,可能遗留认知功能损害、行为问题、癫痫等后遗症。本研究中1例头颅MRI表现为类似“脑白质营养不良”样改变的患儿经小剂量激素维持及每月IVIG,随访1年9个月,未出现复发,未遗留神经系统后遗症,复查头颅MRI 病灶明显吸收(图1 J),但仍需继续延长随访时间进一步观察。
综上所述,MOG抗体相关的ADEM 5岁以下儿童多见,脑病及抽搐为最常见临床表现,急性期大剂量IVMP 联合IVIG 治疗反应好,总体预后较好,部分可复发,缓解期小剂量激素维持配合每月IVIG可能可减少复发。
猜你喜欢
湘潮(上半月)(2022年8期)2022-12-12 03:45:28
昆明医科大学学报(2021年4期)2021-07-23 01:21:32
现代畜牧科技(2021年3期)2021-07-21 08:42:14
大观(2018年8期)2018-01-23 18:02:37
河北医学(2016年5期)2016-12-01 03:58:56
实用临床医学(2016年8期)2016-06-07 01:28:23
扬子江(2016年1期)2016-05-19 23:29:21
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:17:02
磁共振成像(2015年2期)2015-12-23 08:52:22
中医研究(2014年6期)2014-03-11 20:29:02
