
不同养殖密度西伯利亚杂交鲟幼鱼转录组分析
2022-05-07 00:59王子予朱敏杰查云飞简少卿赵大显
江西农业大学学报 2022年2期
王子予,程 超,朱敏杰,肖 敏,查云飞,简少卿,赵大显*
(1.南昌大学 生命科学学院/江西省水产动物资源与利用重点实验室,江西 南昌 330031;2.江西省吉安市井冈山农业科技园,江西 吉安 343000;3.井冈山沐云特种动物养殖有限公司,江西 吉安 343000)
【研究意义】西伯利亚杂交鲟是以西伯利亚鲟为母本、施氏鲟为父本杂交培育出来的一个养殖品种[1],它具有生长速度快、抗病能力强、耐运输等特点,因此在生产实践中深受广大养殖户的欢迎。但随着集约化养殖的发展,为了追求更高的利润,养殖户往往通过增加养殖密度来提高养殖产量,养殖密度是一个复杂且关键的环境影响因子,适当的提高养殖密度是可以达到增加养殖产量的目的,但盲目增加养殖密度容易导致水质恶化以及种内对饵料和空间的竞争,进而导致鱼类生长水平、存活率以及抗病能力的降低。养殖密度也会造成个体生长的差异,且随着养殖密度的增加,差异的趋势越明显[2],因此,研究养殖密度对西伯利亚杂交鲟生长性能的影响及其作用的分子机制对该品种的健康养殖显得尤为重要。【前人研究进展】高通量测序技术也被称为下一代测序(NGS)技术,近些年有一种低成本的NGS 技术受到追捧,它可以用来分析动态转录组,该技术即为RNA 测序(RNA-Seq)。RNA-Seq在硬骨鱼类的转录组学研究中能够更好的帮助了解鱼类的许多生物学过程,如生长发育、适应性进化、免疫反应以及应激反应[3]。李永娟[6]利用RNA-Seq 技术分析了热应激条件下虹鳟肝脏和头肾组织转录组数据,获得了2 种组织共有差异基因36 个,发现了虹鳟热适应相关的调控通路主要为内质网的蛋白加工过程和免疫相关的“NOD 样受体信号通路”,为进一步揭示虹鳟热应激反应的分子调控机制提供了科学数据;张明洋等[7]利用RNA-Seq 对类志贺邻单胞菌感染杂交鲟后的肠道组织进行了转录组分析,共得到差异表达基因13 542 个,其中上调基因9 774 个,下调基因为3 768 个,并筛选可能影响肠道黏膜免疫的相关通路和基因,为类志贺邻单胞菌感染杂交鲟诱导肠道黏膜免疫应答反应的分子机制奠定了基础;李营等[8]利用RNA-Seq 技术对人工养殖2 龄施氏鲟(Acipenser schrenckiiBrandt)精巢与卵巢进行转录组测序分析,获得了雌雄性腺中共有19 690 个差异表达基因转录本,并发现了4 条与卵巢发育相关的通路,为研究鲟性腺分化和发育机制提供了基础。因此,RNA-Seq 技术在冷水性鱼类差异基因筛选和候选基因发掘上发挥重要功能。【本研究切入点】本研究结合生产实践,应用RNA-Seq技术分析不同养殖密度条件西伯利亚杂交鲟幼鱼转录组信息,筛选获得与生长应激相关的基因和调控通路,为井冈山地区西伯利亚杂交鲟健康养殖制定适宜的养殖密度提供理论依据。【拟解决的关键问题】从分子水平阐明养殖密度对西伯利亚杂交鲟幼鱼生长性能影响机制,为井冈山地区鲟鱼流水健康养殖提供科学参考数据。
1 材料和方法
1.1 试验条件
研究对象为西伯利亚杂交鲟,试验用鱼选自同一批次、规格一致、健康无损。养殖实验在井冈山沐云特种动物养殖有限公司鲟鱼养殖基地进行,实验池为9 个四边形室外水泥池,单个水泥池占地面积为28 m2,水深维持在40 cm 左右。流水池主要由进水口、鱼池、出水口组成,每个进水口均为同一侧流向,且3 个部位之间都有30 cm 左右的高度差,鱼池的进、出水口均设置拦网,防止杂质进入、水质遭到污染以及可以有效的防止幼鱼的逃逸。根据连通器的原理设计水交换系统。水源为井冈山山泉水,水体清澈无污染,透明度高,符合《中华人民共和国渔业水质标准》(GB 11607—1989)。
1.2 试验设计
试验鱼初始体质量为(7.89±1.45)g/尾,初始体长为(10.41±0.70)cm/尾。随机分配到9个相同地点养殖池中。本实验设计了3个密度,即低密度(LD)、中密度(MD)、高密度(HD),分别为59 尾/m²、88 尾/m²、118 尾/m²,比例为2∶3∶4。每个密度设置3 个平行组。按照试验设计低密度池每池投放约为1 650 尾,中密度池投放约为2 475 尾,高密度投放约为3 300 尾。养殖试验时间从2019年7月下旬至10月下旬,试验持续90 d。
1.3 饲养管理
试验期间采用饱食投喂法投喂饲料,每天06:00,12:00,18:00 投喂鲟鱼配合饲料。日投喂量为鱼体质量的1%~3%,根据鱼体质量变化,适时调整饲料的规格。每天观察鱼类的摄食情况,对投喂量实施调整。养殖期间,进水口水流流量为100 cm3/s。每天巡塘3 次,观察鱼类状况,定期检测水质变化,养殖期间水质情况见表1。
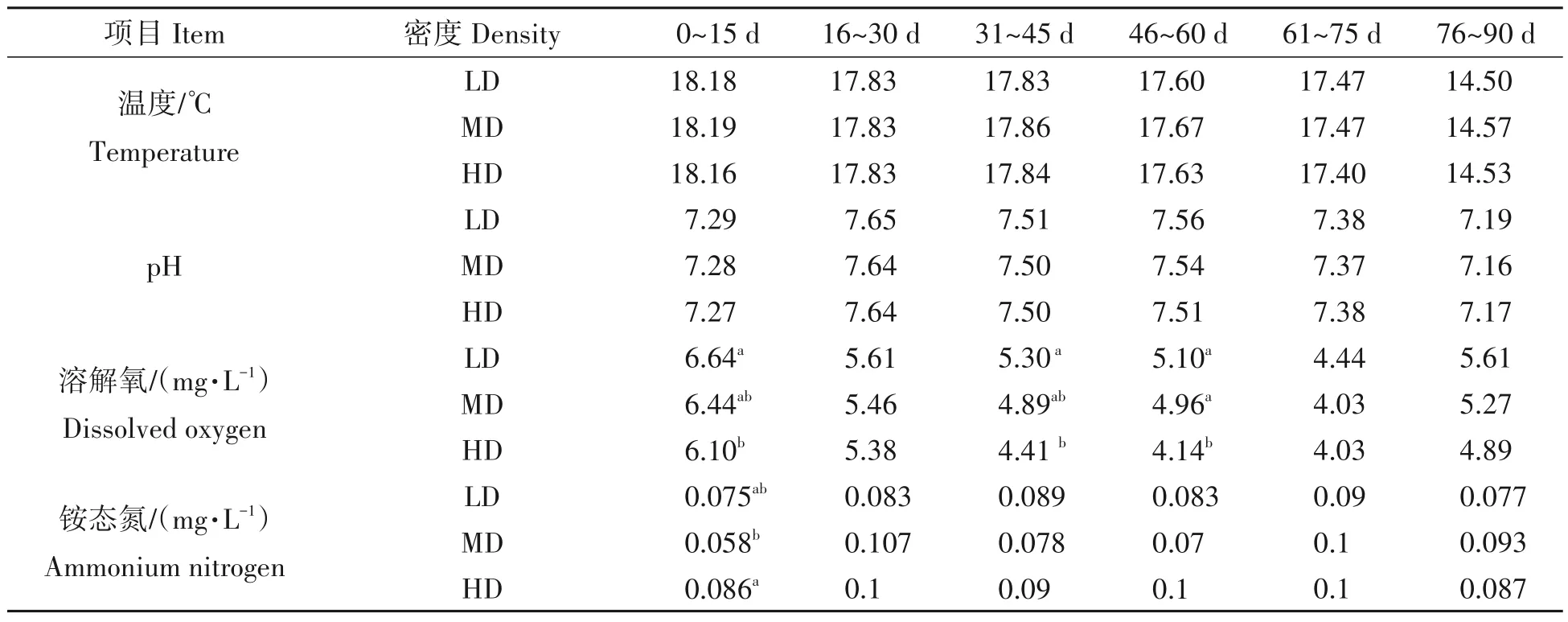
表1 不同密度组养殖水质变化Tab.1 Changes of aquaculture water quality in different density groups
1.4 样品采集
于养殖实验的第90天采集鲟鱼肌肉样本。从每个养殖池中随机捞取10尾鱼进行采样。解剖前,将鱼放入100 mg/L 的MS-222 中麻醉,取背部肌肉组织样品快速放入液氮罐中带回实验室保存在-80 ℃冰箱中用于转录组的测序。
1.5 RNA-Seq测序
1.5.1 总RNA 的提取与质量检测 整个RNA 提取及操作过程使用的枪头和离心管均经过灭菌,实验器具经过0.1%DEPC 水浸泡、高温高压灭菌。从冻存管中取出100 mg组织样品,放入匀浆仪(Servicebio,武汉,中国)研磨至无组织块,按照Trizol法提取总RNA。
使用Nanodrop 2000(Thermo,USA)检测RNA 浓度及纯度:OD260/280值应在1.8~2.2 范围内,OD260/230值范围为1.5~1.8。RNA 完整性检测:配制1%的琼脂糖凝胶,电泳检测总RNA 的质量。将浓度过高的RNA进行适当比例的稀释。
1.5.2 cDNA 文库的构建和Illumina 测序 使用NEBNext UltraTM RNA Library Prep Ki(tNEB,USA)试剂盒生成测序文库并对文库质量进行评价,合格后送至北京百迈客生物科技有限公司(BMK,北京,中国)完成转录组测序工作。。
1.6 测序数据处理与分析
1.6.1 测序数据的产出和质量控制 使用高通量测序平台对cDNA文库进行测序,产出大量的高质量原始数据(raw data),然后通过Perl 程序进行处理获得清洁数据(clean reads),同时计算clean data 的Q20、Q30、GC含量和序列重复水平。
1.6.2 转录组数据和参考基因组序列比对 原始数据经过处理后成为clean data,利用Hisat2 软件将clean data 比对到参考基因组上,然后利用StringTie 把比对上的读长进行组装和定量。本研究选用的小体鲟参考基因组(https://www.ncbi.nlm.nih.gov/genome/?term=acipenser+)。
1.6.3 新基因的挖掘 将拼接好的序列与原有的基因组注释信息比对,探寻原本未被注释的转录区域,发掘该物种新的转录本和新基因,达到补充和完善基因组注释信息的目的。
1.6.4 基因功能的注释 运用BLAST 软件将新基因与数据库比对进行注释,使用以下数据库进行基因功能的注释:Nr、Nt、Pfam、KOG/COG、Swiss Prot、KO、GO。
1.6.5 基因表达水平的定量 为了让片段更真实的反映转录本表达水平,需要将样品中的比对上基因组的读长数目和转录本长度进行统一化处理。采用FPKM作为衡量转录本表达水平高低的指标。
1.6.6 差异表达分析 皮尔逊相关系数r 作为评价生物学重复相关性的评估指标,DESeq2 软件分析样品间的差异表达。参考Lau 等[9]将Fold Change≥1.5 且Pvalue<0.01 作为筛选标准。通过对差异显著性P值进行校正得到错误发生率(FDR)。FDR 作为差异表达基因筛选的关键指标。将3 个比较组的差异基因进行维恩图的绘制。
1.6.7 差异表达基因功主要的GO 富集分析 GO 数据库是一个结构化的标准生物学注释系统。GO 注释系统包括3个主要分支:生物学过程(BP),分子功能(MF)和细胞组分(CC)。
1.6.8 差异表达基因KEGG 通路富集分析 以KEGG 数据库中通路为单位,与整个基因组相比,差异表达基因显著富集的通路。选取显著性q值最小的前20个通路进行分析。
1.7 qRT-PCR验证
从3 个密度组(高、中、低密度组),每个密度组中取3 个平行样,共9 个样本进行qRT-PCR 验证。选取转录组文库筛选获得的差异倍数大的基因,包括与生长和应激相关基因水通道蛋白-4(AQP4)、类丝氨酸蛋白酶抑制剂H1(SERPINH1-like)、磷脂酰肌醇3-激酶调节亚基(PIK3R1)、丙酮酸激酶(PKM)、转化生长因子β-2(TGFB2)及与代谢相关的S-腺苷甲硫氨酸合成酶(MAT2a)、核苷二磷酸激酶2(NDKA2)、L-乳酸脱氢酶A(LDHA)、葡萄糖磷酸变位酶1(PGM1)进行实时定量PCR 检测,根据转录组获得序列使用primei Premier 5.0 软件设计引物(表2)并由生工生物工程(上海)股份有限公司合成。荧光定量PCR 反应程序为:95 ℃下预变性10 min,95 ℃下变性15 s,退火温度60 ℃下反应60 s,72 ℃下延伸30 s,共进行40 个循环数,熔解的反应条件为65~95 ℃,读板30 s 记录荧光量。所有的qRT-PCR 重复3 次。
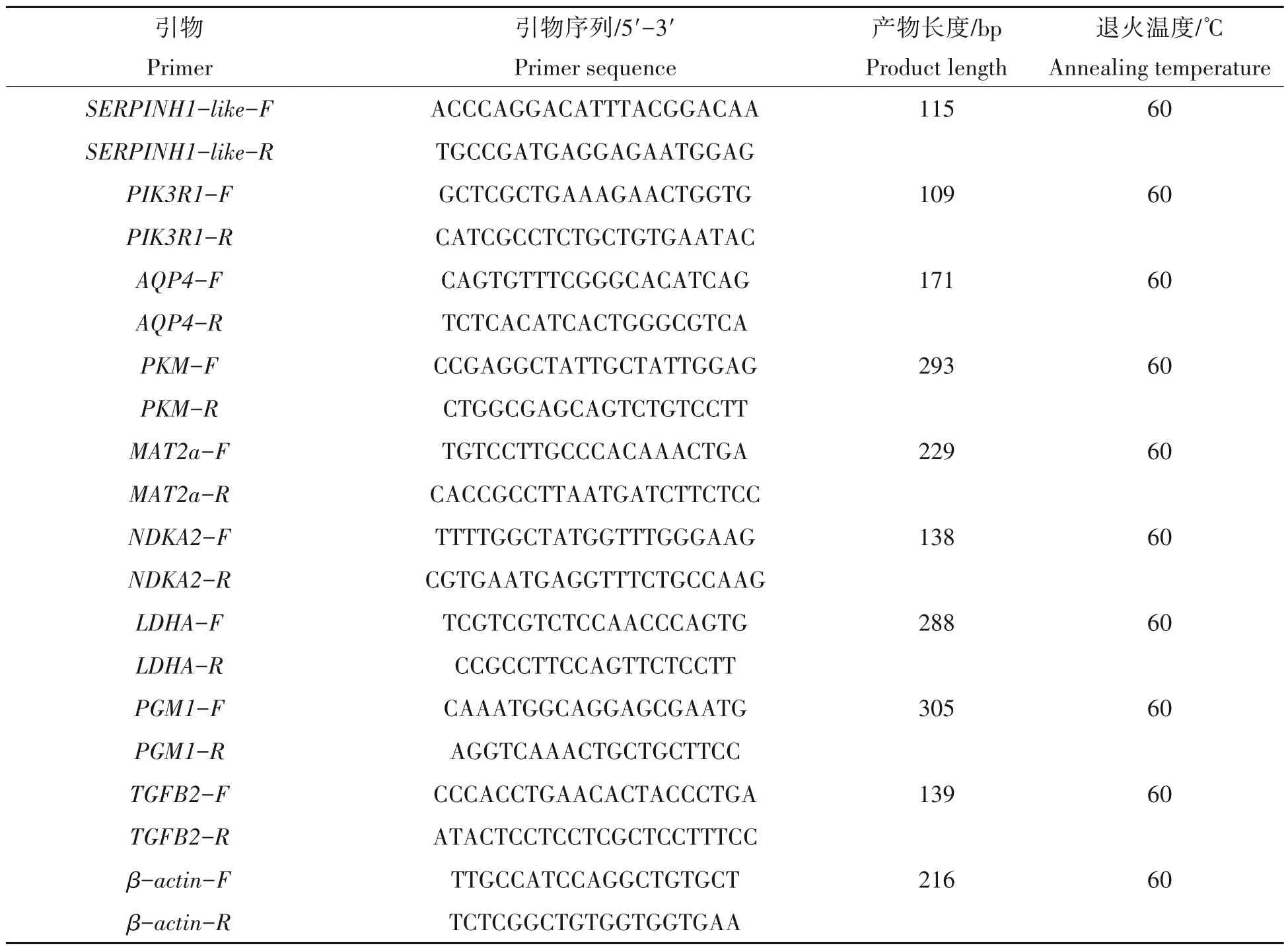
表2 实时荧光定量PCR引物Tab.2 Primers for real-time quantitative PCR
1.8 数据处理
以β-actin为内参基因对得到的各样本的Ct 值做均一化处理,使用2-ΔΔCT方法进行相关基因定量,采用软件SPSS 21.0进行显著性差异分析,Excel 2019进行绘图。
2 结果与分析
2.1 测序结果的产出及质控
试验完成3个密度组(高、中、低密度组),每个密度组取3个平行样,共9个样本进行转录组测序。此次转录组测序共得到60.70 Gb 的clean data,各样品的clean data 均达到6.30 Gb,Q20 和Q30 碱基百分比在97.45%和93.42%以上。GC 含量所占比例平均为低密度组为50.47%、中密度组为50.59%、高密度组50.42%。Clean reads 平均为低密度组23.60×106,中密度组21.91×106,高密度组22.20×106。样品的测序数据见统计表3。
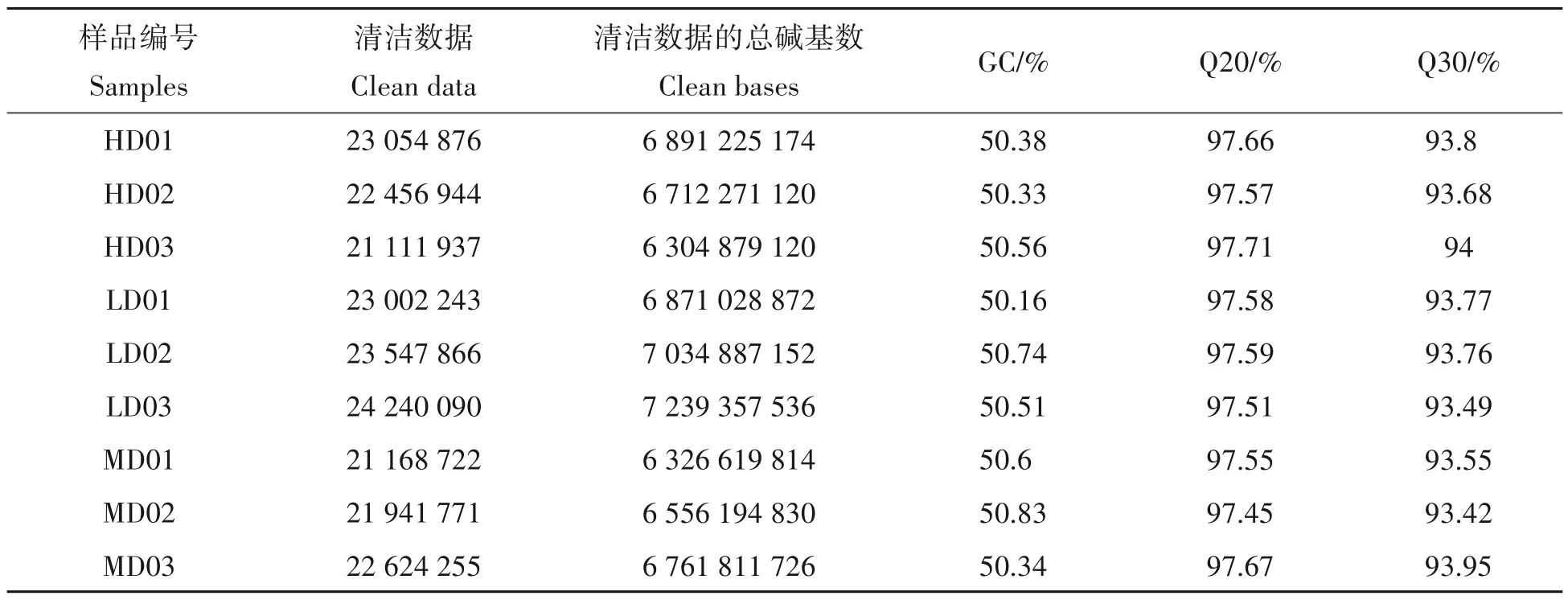
表3 测序数据统计Tab.3 Statistical table of sequencing data
2.2 转录组数据与参考基因组比对
将本试验转录组clean reads 与小体鲟参考基因组进行比对发现,各样品的reads 与参考基因组的比对效率在72.64%~74.38%,比对到基因组唯一位置的reads比例在62.58~63.96%,比对到基因组多个位置的reads 在9.43%~10.42%,比对效果大于70%(表4)。结果表明,可以用小体鲟基因组作为参考基因组进行后续分析。
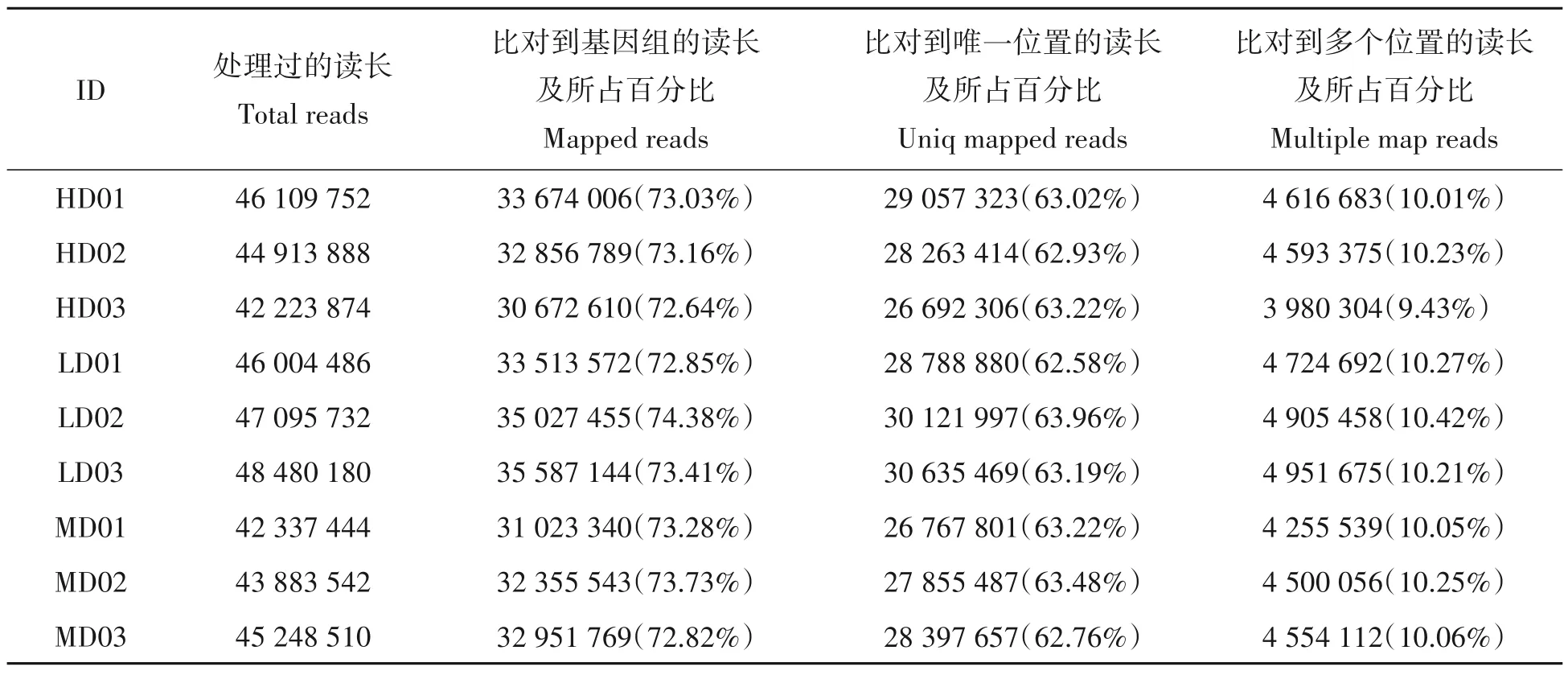
表4 测序数据与小体鲟参考基因组比对结果统计Tab.4 Comparison of sequencing data with the reference genome of small-body sturgeon
2.3 新基因的挖掘与分析
基于小体鲟基因组序列作为参考基因组,共发掘15 549 个新基因。将新基因与数据库比对后进行注释,最终得到各数据库注释的新基因数量统计见表5。结果发现,共12 636 个基因得到注释,2 913 个基因未得到注释,其中COG 数据库注释到2 264个新基因,GO 数据库注释到8 527个新基因,KEGG 数据库注释到8 086个新基因。
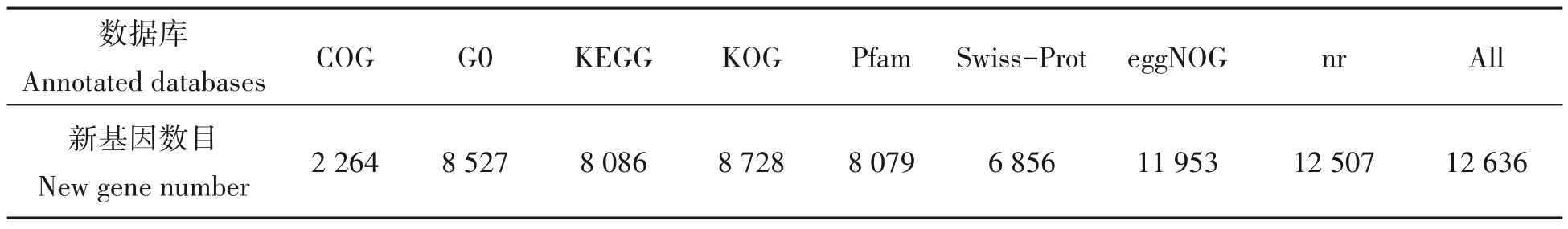
表5 新基因功能注释结果统计Tab.5 New gene function annotation result statistics
2.4 差异基因统计分析
对注释的基因进行差异表达分析发现,LD vs HD 比较组出现235 个差异基因(DEGs),其中108 个DEGs 基因表达上调,127 个下调;LD vs MD 比较组有205 个DEGs,其中111 个基因表达上调,94 个下调;MD vs HD比较组有274个DEGs,其中120个基因表达上调,154个下调(表6)。

表6 差异表达基因数目统计表Tab.6 Statistical table of number of differentially expressed genes
将每组差异基因按照要求进行维恩图绘制发现,总计611 个DEGs,LD vs HD 和LD vs MD 比较组有33 个共有的DEGs;LD vs HD 和MD vs HD 比较组有40 个共有的DEGs;LD vs MD 和MD vs HD 比较组有31个共有的DEGs;只有1个DEG是3个比较组共同有的(图1)。
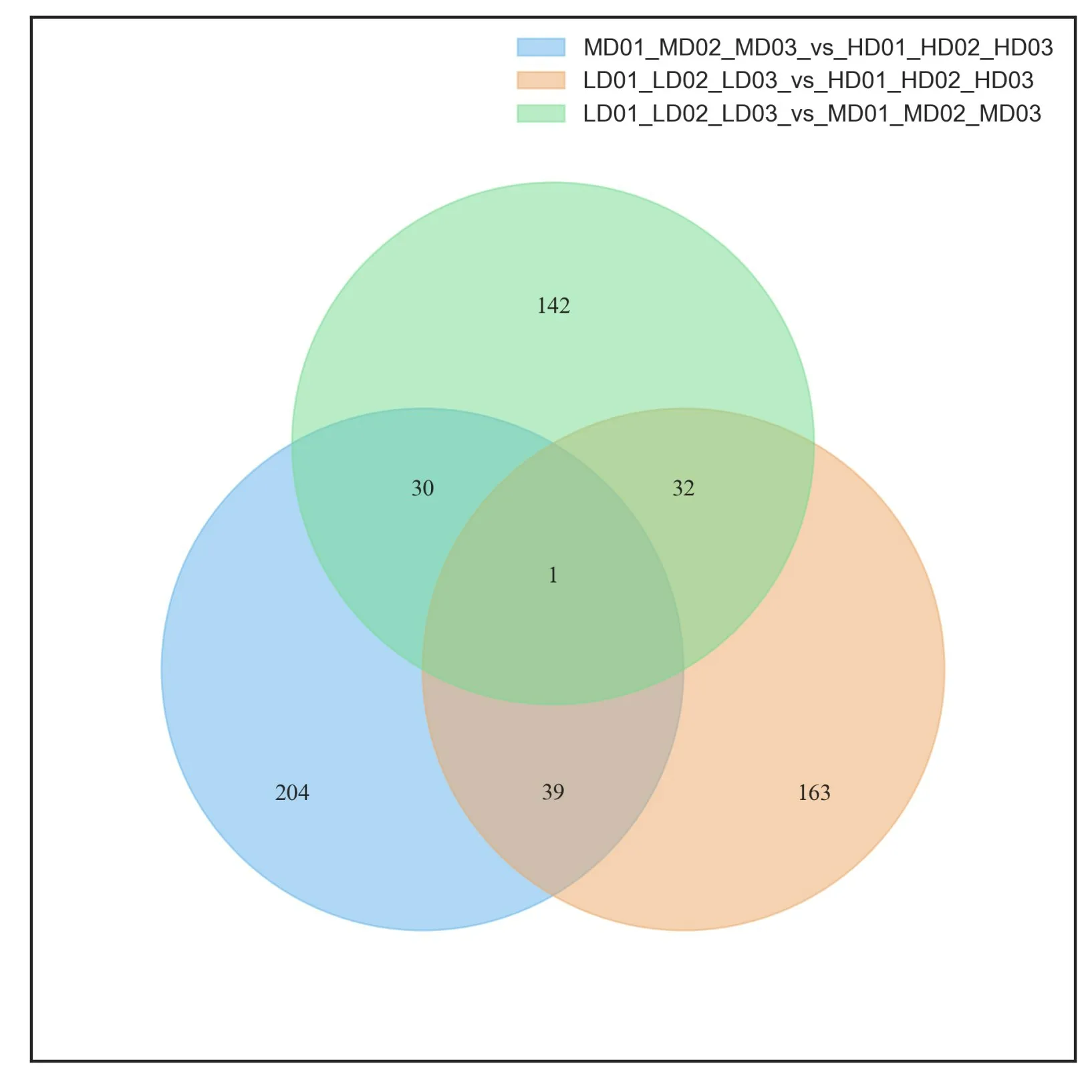
图1 差异表达基因维恩分析Fig.1 Venn diagram of differentially expressed genes
2.5 差异表达基因GO富集分析
对上述DEGs进行GO分类发现。LD vs HD比较组,共有148个DEGs获得了GO注释信息,其中包括62个上调和86个下调的DEGs,归类到GO三大本体(BP、CC、MF)的59个子类别中。由图2A可知,BP主要由代谢过程、细胞过程、单有机体过程、生物调控和对刺激的反应组成;CC 主要由细胞、膜、膜部件、细胞组分和细胞器组成;MF 主要由催化活性和结合组成。在BP 中GTPase 活性的正向调、DNA 介导的转位和DNA整合富集最显著;在CC中,肌球蛋白复合物富集最显著;在MF中,ATP结合、连接酶活性、锌离子结合和肌动蛋白结合。
LD vs MD比较组,共有124个DEGs获得了GO注释信息,其中包括71个上调和53个下调的DEGs,由图2B可知,代谢过程、细胞过程、单有机体过程和生物调控占据着BP大部分;CC主要由细胞、膜、膜部件、细胞组分和细胞器组成;MF主要由催化活性和结合这两个子类别组成。在BP中,GTPase活性的正向调节、DNA整合、整合素介导的信号通路和甘油三酯生物合成过程富集最显著;在CC中,肌球蛋白复合物富集最显著;在MF中,ATP结、连接酶活性、锌离子结合、肌动蛋白结合和GTPase激活剂活性富集最显著。
MD vs HD 比较组,共有173 个DEGs 获得了GO 注释信息,其中包括78 个上调和95 个下调的DEGs,由图2C可知,代谢过程、细胞过程、单有机体过程和生物调控均为BP主要组成;CC主要由细胞、膜、膜部件、细胞组分、细胞器和细胞器部分组成;MF 主要由催化活性和结合组成。在BP中,整合素介导的信号通路富集最显著;在CC中,肌球蛋白复合物和细胞骨架富集最显著;在MF中,ATP结合、连接酶活性、锌离子结合、和肌动蛋白结合富集最显著。
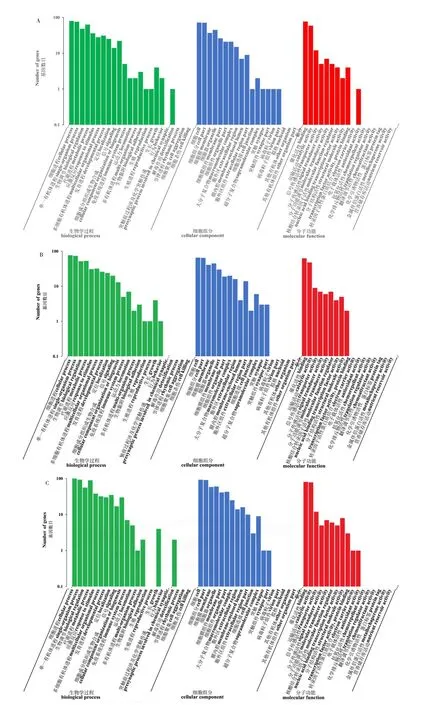
图2 不同养殖密度组别间差异基因注释信息Fig.2 Annotation information of differential genes between groups of different breeding densities
2.6 差异表达基因KEGG通路富集分析
对不同组别差别基因进行KEEG 富集分析发现,LD vs HD 组中,63个DEGs定位到62 个特定的KEGG 代谢途径,主要显著富集的通路为糖胺聚糖生物合成-硫酸乙酰肝素/肝素(3 个DEGs)、半胱氨酸与蛋氨酸代谢通路(3 个DEGs)(图3A);LD vs MD 组中,66 个DEGs 定位到107 个特定的KEGG 代谢途径,主要富集的通路为丙氨酸、天冬氨酸和谷氨酸代谢(6 个DEGs)、吞噬体(10 个DEGs)和mTOR 信号通路(7 个DEGs)(图3B);MD vs HD 分组中,84 个DEGs 定位到89 个特定代谢途径,主要富集在糖酵解/糖异生(10 个DEGs)、半乳糖代谢(7个DEGs)、戊糖磷酸途径(6 个DEGs)、氨基酸的生物合成(8 个DEGs)这4个通路中(图3C)。
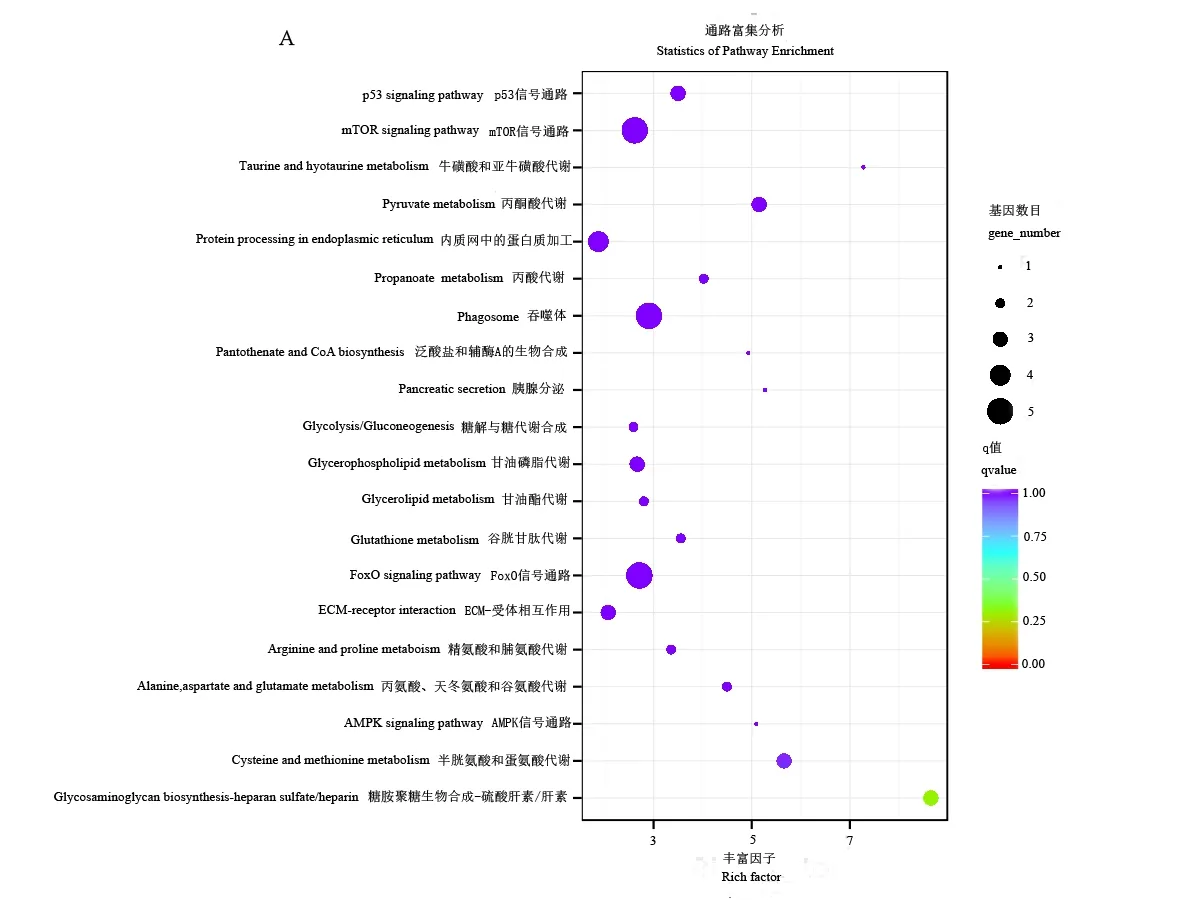
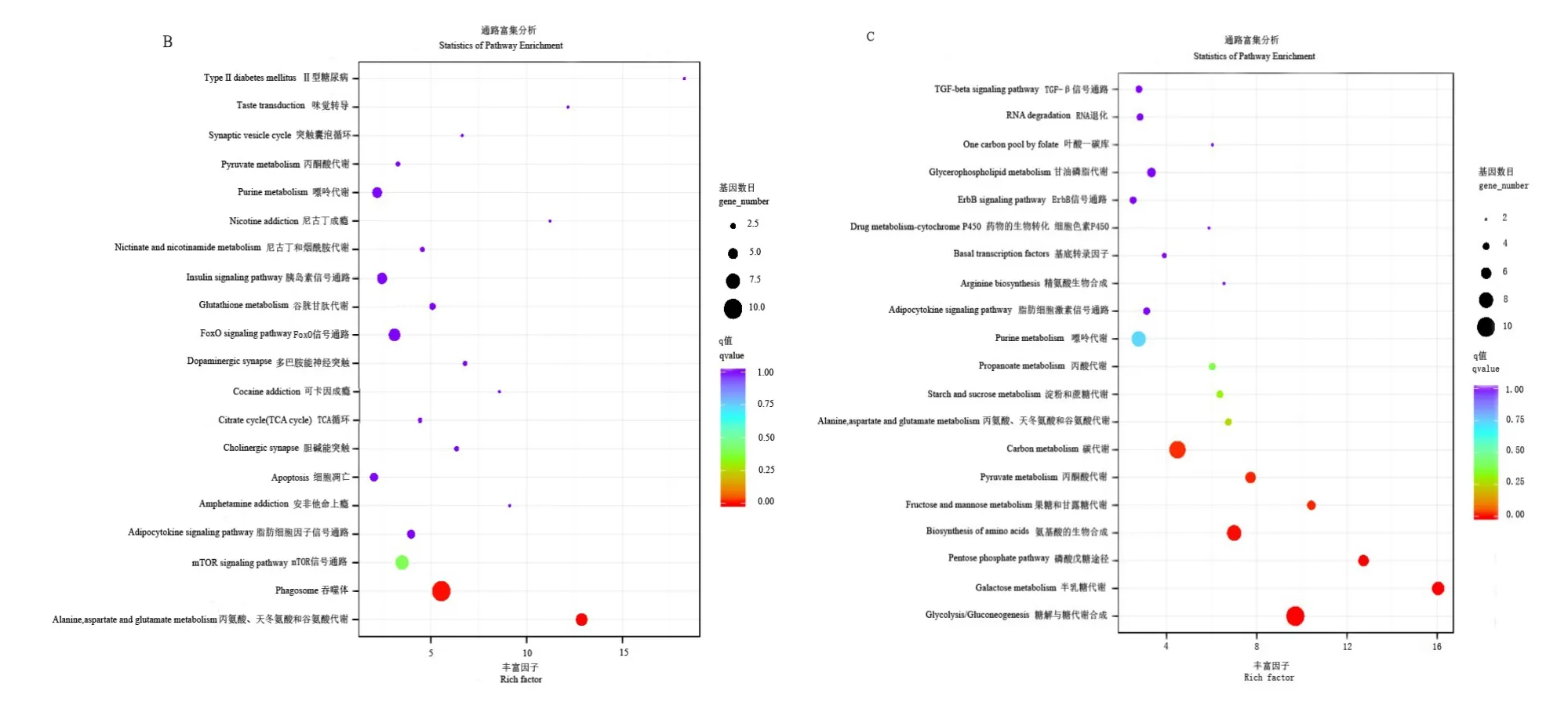
图3 不同密度组差异表达基因KEGG通路富集散点Fig.3 KEGG pathway rich hubs of differentially expressed genes in different density groups
2.7 差异基因中与生长及应激相关基因的筛选与分析
选择p 值小且差异倍数达的差异表达基因,并根据相关文献报道进行筛选。从LD vs HD 分组中筛选获得生长激素受体(GHR)、转化生长因子β -2(TGB-beta2—like)和WNT1 诱导信号通路蛋白1(WISP1)、类丝氨酸蛋白酶抑制剂H1(SERPINH1-like)以及细胞外酪氨酸蛋白激酶PKDCC(Pkdcc)等与生长相关的DEGs及与应激相关的热休克蛋白(HSP70)等;从LD vs MD 组中筛选获得类胰岛素样生长因子(IGF-1)、肌肉骨骼胚胎核蛋白1(MUSTN1)、整合素α-5(ITGA5)以及热休克蛋白90(HSP90);从MD vs HD 分组中筛选获得α-1,6-甘露糖基转移酶(ALG12)和转化生长因子β-2(TGB-beta2—like)、7-脱氢胆固醇还原酶亚型X2(Dhcr7-X2)、成肌细胞测定蛋白1(MYOD1)、丙酮酸激酶11(PKM)、磷脂酰肌醇3-激酶调节亚基(PIK3R1)以及与应激相关的DnaJ 同源亚家族C 第7 成员(DNAJC7)、水通道蛋白4(AQP4)、S-腺苷甲硫氨酸合成酶(MAT2a)、核苷二磷酸脱氢酶2(NDKA2)、L-乳酸脱氢酶A(LDHA)、葡萄糖磷酸变位酶1(PGM1)等(表7)。
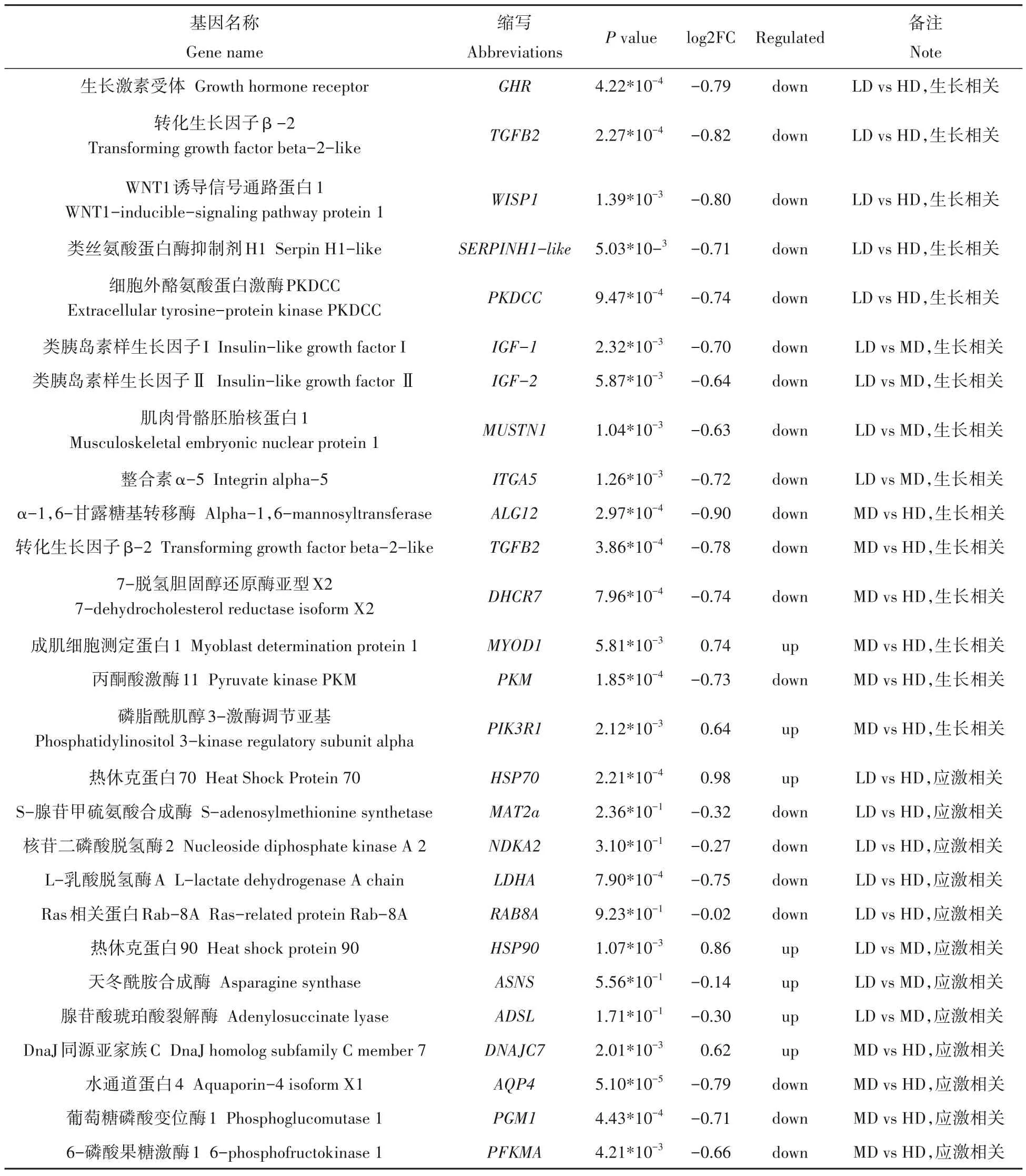
表7 差异基因中生长和应激相关基因的筛选Tab.7 Screening of growth and stress-related genes from differential genes
2.8 qRT-PCR验证
选取9个筛选获得的差异倍数大的基因进行qRT-PCR 验证,结果发现,RNA-seq中与生长和应激相关的水通道蛋白-4(AQP4)、类丝氨酸蛋白酶抑制剂H1(SERPINH1-like)、磷脂酰肌醇3-激酶调节亚基(PIK3R1)、丙酮酸激酶(PKM)、转化生长因子β-2(TGFB2)及与代谢相关的S-腺苷甲硫氨酸合成酶(MAT2a)、核苷二磷酸激酶2(NDKA2)、L-乳酸脱氢酶A(LDHA)、葡萄糖磷酸变位酶1(PGM1)在三个密度组间的变化趋势和qRT-PCR 结果相一致(图4),说明转录组中筛选获得的差异基因表达结果可靠,可以用于后续的基因功能研究。
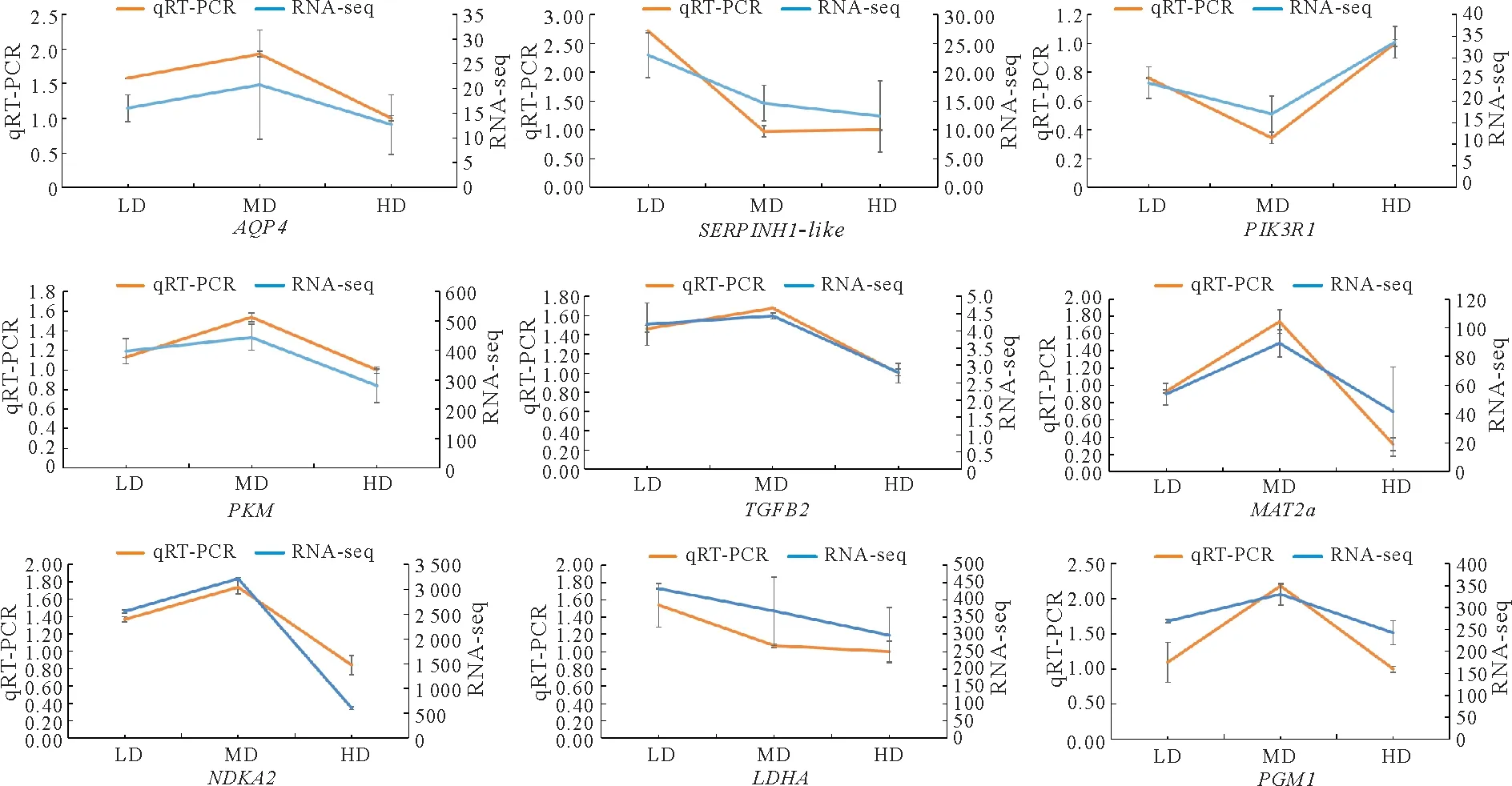
图4 qRT-PCR验证RNA-seq结果Fig.4 RNA-Seq results were verified by qRT-PCR
3 讨论
不同的密度会导致鱼类个体生长产生差异,随着养殖密度的增加,差异越明显。研究认为高密度环境下的鱼类,受到一系列复杂因素的作用,最终导致鱼体生长速度减缓。本试验对3个养殖密度,每个养殖密度3 个生物学重复,共9 个样品进行RNA-seq,共得到60.70 Gb clean data,Q20 和Q30 碱基百分比在97.45%和93.42%以上,将转录数据与小体鲟基因组进行比对,比对效率在72.64%~74.38%(核对2.2 转录组数据与参考基因组比对)。根据转录组与参考基因组的比对,共发掘新基因15 549 个,将新基因与数据库进行比对注释,其中12 636 个基因得到注释,2 913 个基因未得到注释。重复组的设置是为了尽可能的消除生物学可变性的影响,对重复性进行评估发现,同一密度之间3个重复组的相关性均在0.907以上(结果未显示),符合要求。
本试验由3 个比较分组构成,分别为:LD vs HD、LD vs MD、MD vs HD,对不同分组的基因表达进行定量,筛选出差异表达基因(DEGs),并进行注释和功能富集分析。结果在LD vs HD 分组得到235 个DEGs,LD vs MD分组得到205个DEGs,MD vs HD分组得到274个DEGs。差异基因GO富集分析,可以发现各比较分组在GO二级分类富集的差异基因个数和类别均不同。其中,LD vs HD分组的差异基因主要富集在GTPase 活性的正向调节、DNA 介导的转位、DNA 整合、肌球蛋白复合物、ATP 结合、连接酶活性、锌离子结合和肌动蛋白结合,LD vs MD 分组的差异基因主要富集在GTPase活性的正向调节、DNA整合、整合素介导的信号通路和甘油三酯生物合成过程、肌球蛋白复合物、ATP 结合、连接酶活性、锌离子结合、肌动蛋白结合和GTPase激活剂活性,MD vs HD 分组的差异基因主要富集在整合素介导的信号通路、肌球蛋白复合物、细胞骨架、ATP结合、连接酶活性、锌离子结合、肌动蛋白结合。结果表明,养殖密度引起的GTPase 活性变化,有助于细胞分化、生长发育以及免疫应答[10-11],不同放养密度引起的肌肉运动活性变化主要体现在肌球蛋白复合物、肌动蛋白结合、ATP结合以及锌离子结合[12]。
通过KEGG 分析发现,LD vs HD 比较组中,主要显著富集的通路为糖胺聚糖生物合成-硫酸乙酰肝素/肝素、半胱氨酸与蛋氨酸代谢通路;LD vs MD 比较组中,主要富集的通路为丙氨酸、天冬氨酸和谷氨酸代谢、吞噬体和mTOR 信号通路;MD vs HD 比较组中,主要富集的通路为糖酵解/糖异生、半乳糖代谢、戊糖磷酸途径、氨基酸的生物合成。在不同的分组中富集的通路不同,可能因为在不同密度下鱼类的生长、生理状况不同,导致影响程度不同。从这些富集通路可以发现,不同养殖密度显著影响了机体基础代谢,这可能是导致鱼类个体出现差异的原因。
结合文献报道,人工筛选获得了27条与生长与应激相关基因。其中,转化生长因子β-2(TGFB2)具有调节细胞生长和分化的功能[13];WNT1 诱导信号通路蛋白1(WISP1)是一种富含半胱氨酸的分泌型母细胞蛋白,调节细胞反应,如细胞生长、分化和生存[14];胰岛素样生长因子(IGF-1、IGF-2)作为GH/IGFs生长轴的主要成员,调节了大部分生长激素的促生长活性,如细胞生长和分化,DNA 和蛋白质的合成记忆脂质和碳水化合物的代谢等[15];HSP70、HSP90作为评价应激反应的生物指标,同时作为分子伴侣能够维持细胞和蛋白稳态[16];水通道蛋白-4(AQP4)介导水的跨膜转运,是中枢神经系统中含量最高的AQP[17];丙酮酸激酶(PKM)促进平滑肌细胞表型转化和新生内膜增生[18];S-腺苷甲硫氨酸(MAT2a)广泛存在于所有组织和体液中,是生物体内极其重要的代谢物质[19];NDKA2参与多种细胞活动,如细胞增殖、生长、黏附和分化等[20];葡萄糖磷酸变位酶1(PGM1)是糖异生过程中的关键酶,参与糖原的分解和合成,在糖代谢过程中起重要的作用[21];与生长相关的SERPINH1-like参与胶原蛋白的正确合成与分泌[22];L-乳酸脱氢酶(LDHA)是一种关键的代谢酶,优先催化丙酮酸转化为乳酸,其活性的变化往往与外源环境的胁迫密切相关[23];磷脂酰肌醇3-激酶调节亚基(PIK3R1)在细胞的生长和增殖中发挥重要作用[24]。
本试验选取9 个筛选获得的差异倍数大的基因进行qRT-PCR 验证,其中与生长和应激相关的水通道蛋白-4(AQP4)、丙酮酸激酶(PKM)、转化生长因子β-2(TGFB2)和与代谢相关的S-腺苷甲硫氨酸合成酶(MAT2a)、核苷二磷酸激酶2(NDKA2)、葡萄糖磷酸变位酶1(PGM1)在MD 组中表达量最高,这表明MD 组对鲟的生长、应激以及代谢更加有利。而与生长相关的类丝氨酸蛋白酶抑制剂H1(SERPINH1-like)和与代谢相关的L-乳酸脱氢酶A(LDHA)在LD 组表达量最高,随着密度的增加其表达量逐渐降低,推测鲟养殖密度会对其生长和代谢过程产生一定程度的影响。在本试验中PIK3R1在MD组的表达量最低,在HD 中的表达量最高,说明低密度更有利于鲟的正常发育生长。试验选取的9 个基因的qRT-PCR 在3 个密度组之间的变化趋势与RNA-seq 的趋势一致,说明该转录组测序数据结果可靠。综上,不同养殖密度对生长、代谢、应激相关基因的表达有不同程度的影响,且在MD 组养殖对鲟的生长、代谢和应激最有利。
研究对不同养殖密度下的西伯利亚杂交鲟鱼幼鱼进行了转录组测序,从生物信息学的角度揭示养殖密度对鱼类生长影响的潜在机制,为后续研究提供了科学参考。
致谢:江西省现代农业产业技术体系专项(JXARS-03)同时对本研究给予了资助,谨致谢意!
猜你喜欢
军事文摘(2022年16期)2022-08-24
今日农业(2022年4期)2022-06-01
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
今日农业(2021年14期)2021-10-14
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
