
The bone marrow niche landscape: a journey through aging, extrinsic and intrinsic stressors in the haemopoietic milieu
2022-05-06 04:29AntonioGiovanniSolimandoAssuntaMelaccioAngeloVaccaRobertoRia
Antonio Giovanni Solimando, Assunta Melaccio, Angelo Vacca, Roberto Ria
Department of Biomedical Sciences and Human Oncology, Section of Internal Medicine and Clinical Oncology, University of Bari Medical School, Bari 70124 Italy.
Abstract Inflammation and its effects in the bone marrow microenvironment represent a paradigmatic condition in which the hematopoietic niche and the immune systems, thought to properly sustain blood cell production and distinguish between friend and foe, can actively sustain a corrupted neighborhood within a chronic aberrant inflamed state.The bone marrow niche hijacks the physiologic hematopoiesis.The interactions between the hematopoietic stem cells and the niche in the bone marrow are critical determinants of quiescence.We examined several approaches to confront the available evidence; three key points emerged, pointing to the chronic inflammation process, especially the chronic infection and systemic inflammatory states, as leading causes of hematopoietic stem cell depletion.Clonal hematopoiesis, defined as a relative expansion of individual clones, is caused by somatic alterations in essential hematopoietic genes, which increase stem cell fitness.Moreover,terminal differentiation plays a significant role in progenitor loss and inflammatory signaling, promoting clonal selection and clonal hematopoiesis conditions.Specific myeloid malignancies as paradigmatic examples are discussed as a condition associated with inflammation, including the 5q- syndrome, Philadelphia negative myeloproliferative neoplasms, and chronic myeloid leukemia.Aging with increased fitness and hematopoietic stem cell attrition, extrinsic stress, enhanced stressor-specific fitness, and intrinsic defect across the hematopoietic process represent the route for novel insights in defective hematopoiesis.The discussion in this review also pointsout that the hematopoietic niches’ inflammatory stimulation may affect differentiation patterns and the function of downstream cells.
Keywords: Hematopoietic niche, immune system, inflammation, tumor microenvironment
INTRODUCTION
A bone marrow niche can be streamlined as a narrow environment that guarantees the preservation and modulation of the cell stemness.A simplified overview of the bone marrow microenvironment can be sketched around the hematopoietic stem cell (HSC), with quiescent characteristics, capable of self-renewal regulated by multiple niches.One niche is classified as endosteal niche, comprising the bone and several multiple different cell types responsible for the maintenance of the bone and the maintenance of the hematopoiesis[1,2].The perivascular niche also includes multiple and different nursing cells regulating the hematopoiesis in direct crosstalk with the megakaryocytes, macrophages, and endothelial cells that act as gatekeepers for the immune cells[3,4].The innervation and the immune cells are actively fueling the neural niche, regulating the hematopoiesis via the mesenchymal stromal cells, which altogether lead to the maintenance of normal hematopoiesis and homeostasis of the blood system[5,6].Moreover, the erythroblastic island represents a fundamental niche where different stages of red blood progenitors mature over time until they become enucleated cells, subject to the phagocytic process[7].
From this standpoint, inflammation, especially in its chronic phase, can be tightly connected to the normal process of aging, being defined as inflammaging[8,9].A subclinical status of inflammation parallels an increase in visceral body fat over time, leading to the recruitment of blood monocytes and polarization to M1 macrophages, which are even more inflammatory, resulting in an increased production of proinflammatory cytokines[10].On the other hand, both the muscle mass and the anti-inflammatory myokines decrease, causing an increased inflammatory state.Collectively, the red marrow converts to fatty marrow, actively taking part in the systemic inflammaging[11,12].Within the bone marrow, a multipotent stem cell-like cell can give rise to osteoblast and adipocytes under normal conditions, being also crucial to regulated hematopoiesis[12]: the state of aging can be mimicked in a mouse model by a high-fat diet, where the multipotent stem cell-like population tends to differentiate into adipocytes, and there is less differentiation into osteoblasts, leading to increased levels of DPP4, which, when inhibited, orchestrates a more regenerative environment[12].A decreased bone healing mirrors the increased fat, being an underlying cause of the defective hematopoiesis and the lack of maintenance of HSC.Thus, chronic inflammation affects the balanced lineage output during the physiologic homeostasis that, in step with aging and disease,leads to an impaired HSC self-renewal, with increased myelopoiesis[13].Chronic inflammation can be considered not only as a chronic inflammatory disease but also as an aging-mediated process.Thus, HSC can prime a decreased lymphopoiesis and reduced erythropoiesis.Furthermore, these niche dysfunctions and stressors (i.e., reactive oxygen species and acquired mutations) can drive clonal hematopoiesis and a malignant transformation[13].The niche dysfunction can be crucial in the development of full-blown leukemia[14].Clonal hematopoiesis in an average individual with unremarkable hemogram and no evidence of hematologic disease is known as clonal hematopoiesis of indeterminate potential (CHIP) and is also known as clonal hematopoiesis of aging, being a common age-related condition[15].
ETIOLOGY AND BIOLOGY OF CLONAL HEMATOPOIESIS
The hematopoietic system, being probably one of the most proliferative tissues in the body, is physiologically correlated with a high mutational rate.DNA-polymerase can lead to mutations in the most proliferative progenitors and not retained in the progeny.Nonetheless, the mutation sometimes happens inthe stem cell compartments, mostly quiescent, and is retained.Thus, it is not unlikely that the lifetime risk of cancers increases in tissue with a high proliferation rate of stem cells, with some malignancies being more common, such as colorectal cancer and basal cell carcinoma, and some relatively infrequent, e.g.,sarcomas[16].Hematological malignancies are characterized by an intermediate rate of dividing stem cells.Nonetheless, it is estimated that one protein-coding mutation every ten years happens in HSC.Given that about 50,000-200,000 HSCs per person are present, by the age of 70, hundreds of somatic mutations are accumulated in the hematopoietic tissue, known to also be mutated in hematological malignancies[17].The vast majority of DNA is represented by a non-coding one, with 1%-3% containing protein-coding sequences, with the acquisition of somatic mutations being a stochastic process.Therefore, most mutations are inconsequential.Nevertheless, the mutation can occasionally alter tumor proto-oncogene, leading to clonal expansion and subsequent development of hematological malignancies[17].Therefore, it is essential to pinpoint that somatic mosaicism is present in most tissues, constituted by inconsequential mutations.Conversely, clonal hematopoiesis represents a tiny portion of somatic mosaicism.Hematological cancers represent a minority of clonal hematopoiesis[15].To better understand somatic mosaicism, it is pivotal to recapitulate the hematopoietic stem cell life cycle and recognize early mosaicism, resulting in a relatively sizable clone and late mosaicism, being harbored by the progeny of the HSC.From a detection standpoint,these mutations can be detected by next-generation sequencing approaches (NGS) or by ultra-deep NGS and whole-genome sequencing approaches (WGS), which have a broad coverage.The sensitivity in detecting most mutations is high with the latter[18,19], not necessarily implying that those patients have clonal hematopoiesis.In the case of mutations affecting tumor suppressors or oncogenes, leading to increased stem cell fitness, an expansion of the HSC, progenitors, and immature cells can happen, due to genetic alterations involving the coding regions.An expansion of clonal cells can be realized[20-22].Limiting the detection analysis to targeted sequencing of the genes frequently mutated in cancer or looking at WGS and WES from the cancer-associated mutations perspective only, the detectability of clonal hematopoiesis is estimated in approximately 10% of elderly subjects[15].
The underlying mechanism of clonal hematopoiesis is relatively simple, with the fraction of clonal cells being represented by the fraction of mutant cells over normal unmutated polyclonal hematopoietic cells.Thus, the fraction of clonal cells can be increased by the enhanced number of expanded mutant cells or by the decrease of normal cells, mostly due to HSC alterations[15,23].
The clonal expansion is related to the increased stem cell fitness and the mutant cells’ expansion, with relative preservation of normal polyclonal hematopoiesis.Aging is the second mechanism by which the enhanced fraction of clonal cells is related to the attrition of HSC in the polyclonal hematopoiesis counterpart[24].Different models have explained clonal hematopoiesis due to increased HSC fitness and genetic driven HSC attrition.The computational approach pioneered the comprehensive explanation,pointing toward a role of mutations in favoring asymmetric self-renewal, where a mutation can also have a negative effect causing a division into differentiated cells and, therefore, the disappearance of the mutated stem cells or a neutral effect when the number of the mutated stem cells remains the same[25].The mathematical model supported this hypothesis by correlating the size of the clone with the age of the individual.The computational results show that mutations with high fitness led to a rapid expansion of HSC clones.Contrariwise, mutations with moderate fitness will expand much more slowly, and mutations with low impact will remain stable or disappear over time[25].Watsonet al.[25]also considered previously published data and described a high correlation between mathematical prediction and the type of mutation,modeling the prediction on several mutations described in clonal hematopoiesis.They also reported that mutations correlated with myelodysplastic syndromes and acute myeloid leukemia are not common in the genome[25].
Functionally, clonal hematopoiesis and stem cell fitness in clinical and biological contexts have been inspired by HSC transplant-derived data, obtained in a context where the cells with self-renewal potential can engraft into the recipient, recapitulating the entire hematopoiesis.In this context, specific mutations or clones with certain mutations may hold a given stem cell fitness, leading to the expansion of a specific clone[26].Screening 1727 donors, Gibsonet al.[26]identified 388 subjects with clonal hematopoiesis, DNMT3 being the most commonly mutated gene followed by TET2 and other mutations.They also addressed the aim of detecting when the graft and the donors both retained the genetic alteration[26].Moreover, DNMT3A R882 was engrafted in all cases while DNMT3A non-R882 was not.Furthermore, DNMT3A R882-positive cases expanded more rapidly in the donors over DNMT3A non-R882 cases[26].Both increased HSC fitness related to enhanced clonal cells and decreased normal cells due to HSC attrition led to a boosted fraction of clonal cells[15].
Moreover, a context-dependent increase in HSC fitness related to cell-extrinsic stress and genotoxic stress also plays a significant role[27].Some mutations under homeostasis can be relatively neutral but confer increased HSC fitness under certain stressors.One of the most detrimental stressors for hematopoiesis is genotoxic stress, representing a model for understanding the consequences of cytotoxic chemotherapy and radiation[27].Boltonet al.[27]described thatDNMT3Amutation is often detected, but they also observed a relative expansion of the clone carrying mutations in DNA-damage response genes such asPPM1D,TP53,andCHEK2.These mutations were more frequently associated with clonal expansion even in a multivariable logistic model adjusting for age, sex, race, and smoking[27].The authors uncovered the radioisotope-based regimen and external-beam radiation to be highly associated with the presence of clonal hematopoiesis as well as cytotoxic chemotherapy, while target therapy and immunotherapy were not;among the types of cytotoxic chemotherapy, topoisomerase inhibitors or platin-derived compounds were associated with increased risk of clonal hematopoiesis[27].
To better understand the effect of cell-extrinsic stress and infer these mechanisms, the TP53-dependent genotoxic stress related to chemotherapy represents an archetypic example to precisely define if the mutated clone is preexistent or consequential to cytotoxic agent exposure[28].Wonget al.[28]reported that TP53-mutated clones are present before chemotherapy is administered, and, during chemotherapy, this clone can expand, acquiring additional mutations, evolving in a therapy-related myeloid neoplasm.McNerneyet al.[29]substantially reviewed these findings by providing a model in which healthy individuals carry TP53 mutations that are generally neutral, but, after radiation or chemotherapy, novel mutations drive the transformation into myeloid malignancies.
The immune attack represents the other example of cell-extrinsic stress, particularly T-cell mediated attack is implicated in the pathophysiology of aplastic anemia and paroxysmal nocturnal hemoglobinuria[30].In these cases, BCOR, BCORL1, and PIGA mutations are likely neutral under homeostasis, but the cells with these alterations can evade the cytotoxic effect of T lymphocytes and expand, particularly after immunosuppressive treatment[31].The role of DNMT3A and ASXL1 in immune evasion is much less elucidated, despite subjects affected by these mutations being at high risk of progression to myelodysplastic syndrome (MDS) and AML[31].However, their primary role seems to be played in inflammation and aging.
Clonal hematopoiesis, aging, and inflammation
An increased prevalence of somatic mutations according to age, mainly involving DNMT3A, TET2, and ASXL1, ensues from dissecting CHIP- and age-related clonal hematopoiesis; moreover, CHIP is associated with a greater risk for inflammatory co-morbidities (i.e., type II diabetes, atherosclerosis, or stroke)[20,21][Figure 1].
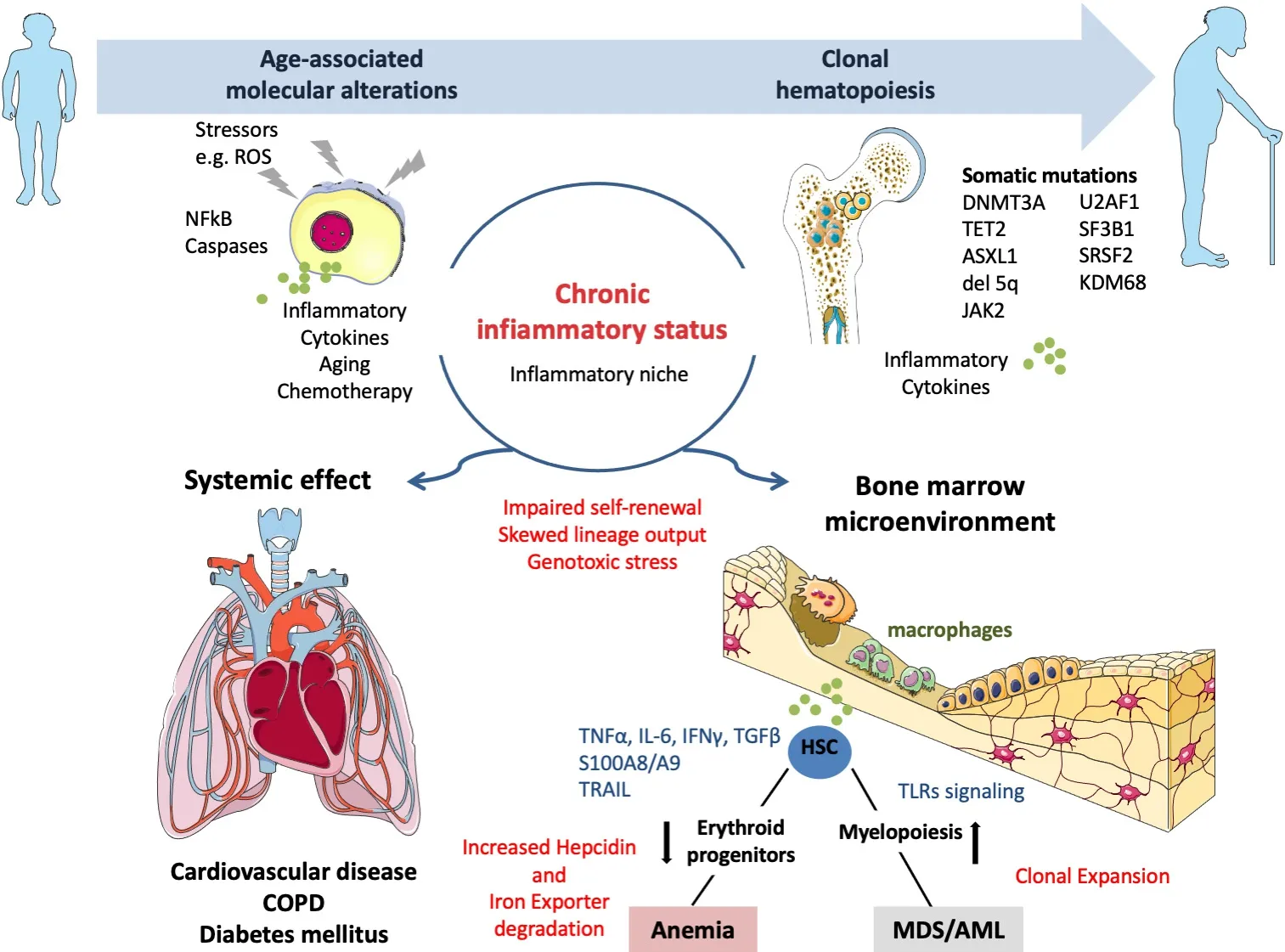
Figure 1.The effects of aging and extrinsic and intrinsic stressors for the bone marrow microenvironment ecosystem: implications for clonal hematopoiesis.ROS: Reactive oxygen species; HSC: hematopoietic stem cell.
CHIP causes inflammatory microenvironments throughout the body since mutations in HSPCs give rise to inflammatory macrophages and neutrophils[32-34].In more detail, TET2, DNMT3A, and JAK2 mutations have been extensively investigated regarding their correlation and role played in inflammatory disease state,atherosclerosis, ischemic heart failure, and thrombosis neutrophils[32-34].Mutations of TET2 and DNMT3A are associated with ischemic heart failure mediated by recruitment of monocytes that at first are circulating in the blood and then can infiltrate the vasculature and the higher release of IL-1β, IL-6, and chemokines fueling an inflammatory state and directly contributing to an inflammatory niche outside the bone marrow[32].Moreover, the JAK2 mutation leads to an increased netosis, which later can increase the thrombus formation in the vasculature of the lung[35].Thus, in aging, a CHIP can occur without being associated with a hematopoietic malignancy; additionally, the subclinical inflammatory status can lead to systemic effects on the cardiovascular system, respiratory system, endocrine glands, and bone marrow[36].Anemia represents the poster child in the sketch of systemic inflammation effects within the bone marrow,where macrophages are central cells in mediating inflammation inside and outside the bone marrow[37].IFN-γ increases myelopoiesis while decreasing the self-renewal of the HSC[38]; conversely, TNF-α is specifically known to inhibit erythropoiesis[39]and influences erythroblast and systemic cytokines, such as interleukin six (IL-6) and IFN-γ.IFN-γ is not the only inflammatory cytokine that can induce hematopoietic stem cell activation[40], since it is now recognized that IFN-α[41], toll-like ligands such as LPS[42], interleukin one (IL-1)[43], and several others can induce stem cell activation.Inflammatory signals, but not the infection itself, trigger changes in stem cell quiescence, self-renewal, and differentiation.Chronic inflammation drivessustained anemia based on inflammatory cytokines throughout the body, affecting macrophages in the bone marrow[44-47].MDS also constitutes a condition associated with cytopenia, particularly in anemia, in which hematopoietic stem and progenitor cells acquire a mutation that leads to a clonal expansion of immature cells; additionally, in the bone marrow, variable grades of dysplasia are present, with various degrees of cytopenia[48].Mathematical models also uncovered the mechanisms of HSC loss, highlighting the relevance of impaired division, increased cell death, displacement, and abnormal differentiation[49].Nonetheless, the stem cells that seem to be lost in pre-clinical models, at least during chronic infections, are primarily damaged by an aberrant terminal differentiation[49].The model is that typically hematopoietic stem cells sit right up close to the CXCL12-abundant reticular cells, but in the setting of IFN-γ stimulation they may upregulate Bst2, which pulls them over to a different part of the niche that E-selectin marks, and this may release them from quiescence and let them divide[50].
Remarkably, inflammation in the bone marrow already primes HSCs for malignancy; in fact, a normal HSC is under the stress of persistent chronic inflammation due to multiple phenomena, such as chronic diseases and aging.Therefore, toll-like receptor (TLR) signaling plays a role in mediating the inflammatory stress,making the HSC more susceptible to acquiring mutations correlated to MDS[48].Higher inflammatory status is also observed, due to a higher secretion of proinflammatory cytokines from the HSC, affecting the microenvironment trapped into a vicious cycle that feeds on itself.
Established animal models based on deficient ribosomal protein S14 (RPS14) phenotype allow studying the level of myelodysplastic anemia, and, while harvesting stem cells from RPS14-deficient mice, they are not able to differentiate into blood cells.The underlying cause for the break in the erythroid differentiation is most likely related to increased levels of S100A8 and S100A9 proteins, both related to the immune response and inflammation via TLR4 and NF-κB activation, being a potential therapeutic target both in MDS and in chronic inflammation.S100A8 expression is found in the erythroblastic island in RPS14-deficient mice,where increased S100A8 also induces P53-dependent differentiation block activation[51].Therefore,inflammation can be a target due to the paradigmatic role of S100A8 as an essential factor for the erythroid differentiation while being a potential therapeutic target in MDS patients with anemia; furthermore,increased S100A8 expression has also been observed in the endosteal and perivascular niche[52].Hence, there is a need for a close investigation of S100A8 expression in an endosteal niche, the mesenchymal component of the endosteal milieu of MDS, and bone marrow failure such as Schwachman-Diamond syndrome[53].By performing immunohistochemistry staining, CD271 highlights the mesenchymal stroma compartment and allows stratifying patients with low-risk MDS according to S100A8 low and high expressors while identifying a worse survival for patients who express high S100A8 levels[53].These individuals have a higher frequency of leukemic evolution, potentially showing that inflammation can represent a trigger in this disease, priming the hematopoietic cells for transformation depending on the microenvironment.
By isolating the stromal cells and co-culturing them with normal HSPCs, upon upregulation of S100A8 on the stromal cells, a decreased hematopoietic support was observed, with downregulation of CXCL12; a lack of quiescence was also obtained, with cell cycle activation and proliferation along with increased apoptosis in HSPCs[53].Based on these pieces of evidence, inflammation represents a target for disease, as exemplified by the expression of S100A8 in the endosteal and perivascular niche, driving a loss of ordinary hematopoietic support and leading to genotoxic stress, sustaining malignant hematopoiesis and priming a more mutagenic environment, in the context of senescence.
The multiple inflammatory targets described affect the environment in the bone marrow, specifically leading to anemia, while the inflammation close to the endosteal and perivascular niche seems to affectnormal hematopoiesis and might also induce malignant transformations[9].The pathomechanism of S100A8/A9 in MDS points towards an inflammasome and pyroptosis driven development of clinically relevant phenotypes[54].S100A9 leads to enhanced oxidative stress while activating the inflammasome,enhancing the proinflammatory signaling that fuels the proliferative and mutagenic effect, and opening a therapeutic window.
DISEASES CAUSING INFLAMMATION IN THE BONE MARROW NICHE
Further studies looking specifically at the different cell types composing the niche focused on myeloproliferative disorders, namely myeloproliferative neoplasms such as JAK2V617Fand TPO drove[55,56],acute myeloid leukemia[57,58], and chronic myeloid leukemia[59].
Inflammation causes “neuropathy” in the bone marrow of multiple malignancies such as acute myeloid leukemia and other myeloproliferative neoplasms.The constitutively active inflammatory status disrupts the interaction of the glial cells with mesenchymal stromal cells, leading to full-blown leukemia, MPN, or bone marrow fibrosis[55,57,59].Thus, dysregulation of the neural niche is an appealing therapeutic target that might affect inflammation; a Beta3-adrenergic agonist, neural-glial protection, and IL-1R antagonist are being studied[55,57,59].Fibrosis in the microenvironment in the bone marrow represents the most massive remodeling of the milieu and recognizes both solid and hematological underlying disease in addition to metabolic disease, immune-related disorders, infections, and aging[60,61].In more detail, a more robust side population of Gli1poscells, responsible for this remodeling of the bone that aligns the bone in normal conditions, can also be found in proximity to the vascular localization[62,63].By inducing fibrosis in animal models, these cells are activated from the usual niche and suddenly are expanded in bone marrow fibrosis.Notably, ablation of Gli1poscells rescues bone marrow fibrosis.
Thus, the underlying processes of inflammation seem to characterize the pre-fibrotic phase, and, in the HSC, there is an increase of inflammatory signaling toward NF-κB signaling, TNF-α, and through the activation of JAK-STAT[64].Interestingly, in the early stages when fibrosis starts, the signature of inflammation as a disease initiator is also present.On RNA sequencing at a single cell level of the nonhematopoietic niche, four different populations have been found: mesenchymal stromal cells (MSC-1/2)and Schwan cell progenitors constituting the neuronal niche, as glial cells, the osteolinear cells, and the vascular cells[64].Focusing on the MSC-1/2, it has been shown that they are functionally reprogrammed in fibrosis, losing their conventional markers profile; in addition, they seem to express fewer MSC markers,ultimately losing their function of hematopoiesis support, and they start making collagen.Therefore, only two of the different populations of cells are responsible for this massive remodeling of the bone marrow through extracellular matrix synthesis, collagens, secretion factors, and neo-angiogenesis[64].In the profile of secreted factors, the spread of S100A8/9 in myeloproliferative neoplasms and myelofibrosis with a massive increase of S100A8 in patient plasma has been reported.This increase could correlate, based on the above,with the deficient phenotype of RPS14 for MDS.In other words, the alterations observed in ribosomal gene expression and translation-related gene expression in HSC of 5q- syndrome are a consequence of the haploinsufficiency of RPS14.These aberrations can compromise the biogenesis of the ribosome and consequent reduction of the translation efficiency of proteins[65].In this frame of thinking, reactive oxygen species can be produced either because of somatic mutations or due to S100A8 or S100A9.As alarmins molecules, they are shed during pyroptosis and tip the balance between immune and inflammatory responses[51,64-66].These events ultimately induce caspase 1 activation, which in turn drives a plethora of downstream consequences such as IL-1β- and IL-18-mediated cell damages[51,64-66].These underlying mechanisms are distinctive of MDS[65]and represent novel, promising Achille’s heels to improve hematopoiesis in myelodysplastic syndromes[51,64-66].
Thus, a biomarker profiling for fibrosis onset can also represent an attractive therapeutic target[64,66].Moreover, the need for timely and niche-oriented treatment can represent a breakthrough.Multiple targets directly treat the underlying disease, such as IL-6 inhibition[67,68].Nonetheless, aging-induced inflammation is a physiological process involving an inflammatory microenvironment and corrupted angiogenesis[69,70].Regarding optimal treatment timing, hemoglobin seems to be a sensitive marker since erythropoiesis is a sensitive process that is mainly impacted by inflammation.Nevertheless, hemoglobin, transferrin, and ferritin concentrations should be reviewed and hopefully be made age-specific[71-73].Cytokine blockade, antiinflammatory therapy, anti-senescence therapies, and specific targets for the hematopoiesismicroenvironment crosstalk are promising approaches[74].Notably, exercise affects the microenvironment and hematopoiesis and reduces cardiovascular risk[75].Those results are supported by pre-clinical data pointing towards a direct role of exercise in decreasing leptin levels.These homeostatic changes halted the vicious cycle between cell quiescence and decreased hematopoietic output of inflammatory leukocytes’hematopoietic output through altered chromatin accessibility of cell cycle genes.Indeed, exercise has been related to increased HSC quiescence, intact niche, and decreased inflammatory cells[75].
Context-dependent increase in HSC fitness: cell-intrinsic germline defect
Inherited bone marrow failure, such as somatic mosaicism, represents a paradigmatic example to understand that the random mutation can involve a group of cells harboring intrinsic germline defects.Some mutations can be detrimental and others neutral, but HSC fitness can also be increased, leading to somatic reversion with partial restoration of normal hematopoiesis[76].The exact mechanism, in this case,can be represented by a direct correction, such as in Dyskeratosis congenital, in which a mutation in the telomerase complex is relatively frequent and somatic mosaicism may lead to the correction of the mutation, restoration of gene function, and restoration of hematopoiesis[77].Indirect correction can also be realized by partial function restoration, such as in Fanconi anemia.Furthermore, as learned from severe congenital neutropenia, CSF3R mutation can lead to an augmented granulopoiesis[78].A fourth example is Shwachman-Diamond syndrome, when TP53 mutation or loss of EIF6 is implicated in bone marrow failure and can lead to partial restoration of hematopoiesis.Unfortunately, some of these mutations also predispose to MDS and AML.Finally, this unravels how these features of the bone marrow neighborhood are linked to molecular mechanisms of corrupted vessel formation, cell adhesion, and aggressive phenotype[34].Crucial mechanisms promoting bone marrow niche corruption due to alteration in the vascular and endosteal also mediate immunosuppression during malignant development and progression[8,79].As is now well known,hematopoiesis grows and evolves through constant crosstalk with the surrounding milieu, and emerging evidence indicates that several gatekeepers and immune-corrupted environments frequently occur simultaneously in response to this crosstalk between the hematopoietic or committed cells and the vascular niche as immune-patrolling checkpoint[4,80].Accordingly, cell-adhesion modulators and strategies combining vascular-directed therapy and immunotherapy seem to hold promise to tip the balance of the bone marrow niche and improve patient outcomes[81-85].
CONCLUSIONS
The survival and commitment of stem cells in the bone marrow are ensured by the hematopoietic niches’microenvironment, namely the osteoblastic and the perivascular.The control mechanisms of hematopoiesis are finely regulated at this level and are based on both direct and indirect intercellular communications,ensuring proper and constant support to stemness.Somatic mosaicism can affect genes crucial in cell function, such as self-renewal, which may lead to increased HSC clonal expansion observed in aging.Chronic inflammation, related to several neoplastic and non-neoplastic diseases, causes genetic and epigenetic changes in the stromal cells and the release of inflammatory cytokines in the microenvironment of stem niches.The switch toward an inflammatory microenvironment causes direct consequences on the regenerative capacity and maturation of the same hematopoietic stem cells affecting the hematopoiesis.Asstated above, the correct treatment of the inflammatory state is of fundamental importance in the therapy of these patients to restore the correct hematopoiesis and physiological homeostasis.The mutations in HSC due to increased stressor-specific fitness can be neutral under homeostasis.Nonetheless, their imposed increased self-renewal under immune attack, inflammation of genotoxic stress, can lead to the expansion of the mutated clone.Finally, in inherited bone marrow failure syndrome, somatic mutations may result in near normalization of the stem cell fitness primarily due to the correction of the underlying HSC defect,leading to partial restoration of normal hematopoiesis.
DECLARATIONS
Authors’ contributions
Conceptualization: Solimando AG, Ria R
Writing: Solimando AG, Melaccio A
Review and editing: Ria R, Vacca A
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by: INNOLABS-Programma Operativo Regionale (POR) Puglia Fondo Europeo di Sviluppo Regionale (FESR)-Fondo Sociale Europeo (FSE) 2014 to 2020 Telemielomedicina/Telemielolab(Vacca A) and by Apulian Regional project: Precision Medicine number 06062019 (Solimando AG).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
 Journal of Cancer Metastasis and Treatment2022年3期
Journal of Cancer Metastasis and Treatment2022年3期
- Journal of Cancer Metastasis and Treatment的其它文章
- Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice
- Diffuse large B-Cell lymphoma: from novel molecular classifications to tailored targeted therapies
- The WHO 2021 thymoma classification: a work in progress
- Chronic activation of MUC1-C in wound repair promotes progression to cancer stem cells
- AUTHOR INSTRUCTIONS