
Diffuse large B-Cell lymphoma: from novel molecular classifications to tailored targeted therapies
2022-05-06 04:29JenniferLueGrzegorzNowakowski
Jennifer K.Lue, Grzegorz S.Nowakowski
1Department of Medicine, Lymphoma Service, Memorial Sloan Kettering Cancer Center, New York, NY 10021, United States
2Department of Medicine, Mayo Clinic, Rochester, Minnesota, MN 55902, United States
Abstract Diffuse large B-cell lymphoma (DLBCL) is a heterogeneous disease comprising multiple genetic subtypes that translates and impacts clinical outcomes after standard chemoimmunotherapy.Our initial understanding of the complex biological subtypes of DLBCL began with the identification of cell of origin (COO), and now has evolved to include even more specific subtypes defined by genetic signatures and mutations.These newer classifications lend themselves to the application of precision-based medicine, allowing us to tailor new treatment platforms that target specific oncogenic drivers in order to improve DLBCL outcomes.Essential to this is the development of genetic assays and tools that are reliable and readily available to assist in the application of these molecular classifications to real-world use.In this review, we discuss the history of DLBCL classification systems and their implication on clinical investigation as well as novel therapeutic options in DLBCL.
Keywords: DLBCL, tumor heterogeneity, activated-B-cell, germinal center, molecular targets
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma (NHL) and is characterized by highly diverse molecular subtypes.Gene expression profiling (GEP) has delineated twomain DLBCL subtypes: germinal center B-cell-like (GCB) and activated B-cell (ABC), with a third group dubbed ‘unclassifiable’ for tumors that are unable to fit discretely into the two former groups[1].These subtypes have differing oncogenic drivers that impact the clinical outcome, with GCB-DLBCLs having a superior overall survival (OS) compared to ABC-DLBCLs[1].More recent genetic classification systems have divided DLBCL into even smaller subgroups[2-6], which also have prognostic implications after standard of care rituximab cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP).In this review, we detail the history of DLBCL classifications, the implications of these new molecular systems, and review recent therapeutic agents for the treatment of DLBCL.
CLASSIFICATION SYSTEMS: IMMUNOHISTOCHEMISTRY TO GENE EXPRESSION PROFILING
Over the past several years, our understanding regarding the immense heterogeneity that lies within DLBCL has led to new molecular classifications based on diverse genetic profiles[1].Understanding the complex molecular background of DLBCL may lead to improved therapeutic approaches in the future.Herein, we describe the evolution of DLBCL taxonomy, starting with cell of origin (COO) designation to more recent systems that further sub-classify DLBCL into smaller hierarchical groups [Table 1].
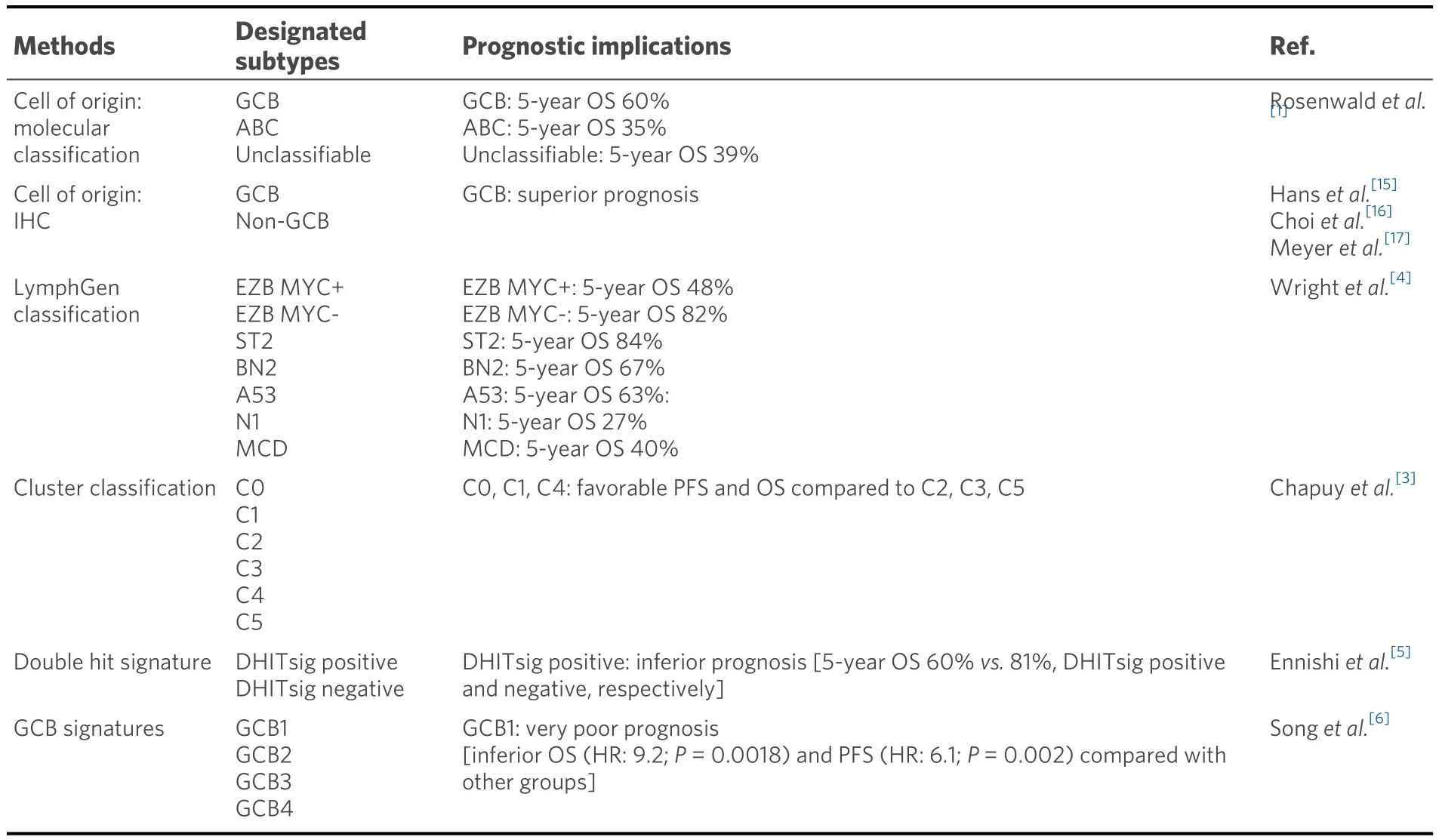
Table 1.Classification methods and prognostic implications
The concept of cell of origin: activated B-cell vs.germinal center Diffuse Large B-cell Lymphoma
The premise of classifying DLBCL into subtypes can date back to studies suggesting that CD10, the common acute lymphoblastic leukemia antigen, could differentiate two classes of DLBCLs[7-10], translating into potential clinical implications based on an expression.Subsequently, using GEP, Alizadeh and colleagues[11]identified two distinct DLBCL subtypes, termed GCB and ABC, both of which are defined by expression patterns that align with different stages of B-cell maturation.The authors concluded that after standard of care treatment, GCB and ABC-DLBCLs are characterized by drastically different OS, with the 5-year OS of GCB-DLBCLs being 76%vs.16% for ABC-DLBCLs.The enhanced understanding of the heterogeneity of DLBCL led to the identification of a third subtype termed “type 3”, or what we have commonly come to know as DLBCL-unclassifiable, which represents a group of tumors that do not discretely fit into ABC or GCB subgroups1.Similarly to what was described by Alizadehet al.[11], and limited by retrospective analysis, this study confirmed that after anthracycline-based chemotherapy, GCB-DLBCL patients demonstrated a superior OS compared to their ABC and unclassifiable counterparts (5-year OS:60%vs.35%vs.39%, respectively).
Another study evaluated 58 DLBCL biopsies using oligonucleotide microarrays (Affymetrix)[12]and identified a 13-gene model that was able to predict dichotomous outcomes: curedvs.fatal/refractory.Using the Affymetrix platform, Shippet al.[12]and colleagues attempted to validate COO genes identified by Alizadehet al.[11]as a surrogate for prognosis after R-CHOP, while acknowledging the limitations that come with this attempt which include: differential technology used by both groups (cDNA arrays/Lymphochip by Alizadehet al.[11]vs.oligionucleotide arrays/Affymetrix by Shippet al.[12]); differing panel of genes probed;varying computational approaches; and lastly, different tumor samples studied.Notably, the two GEP arrays developed by the separate groups shared 90 overlapping COO signature genes.As such, this 90 gene panel was used to cluster DLBCL samples into ABCvs.GCB-DLBCL and was able to confirm the inferior outcome of ABC-DLBCL tumors in the Alizadehet al.[11]samples (Lymphochip).However, no relationship between COO and clinical outcome was observed in the 58 DLBCL samples used in Shippet al.[12]’s(Affymetrix) developmental analysis.In an attempt to reconcile these discrepancies, another classification method was developed not only to distinguish COO, but also to estimate the likelihood that a tumor falls within a particular subgroup[13].Applying this classification system to both the Lymphochip[1]and Affymetrix[12]datasets, tumor samples identified as GCB had a clear survival advantage compared to those classified as ABC[13].Thus, we can conclude several principles from these early GEP studies: (1) the method by which GEP is established can impact interpretability and applicability to patient samples/outcomes; (2)reproducibility across laboratories is key for clinical applicability; and (3) intrinsic to the reproducibility is a consensus on what set of genes represent the optimal way to designate COO.
In another attempt to understand the complex phenotype of DLBCL, using whole genome arrays and three clustering algorithms (hierarchical clustering, self-organizing maps, and probabilistic clustering), Monti and colleagues elucidated three unique DLBCL clusters: (1) Oxidative Phosphorylation (“Ox-Phos”)characterized by an increased expression of NADH complex and cytochrome c/cytochrome c oxidase complex, and BFL-1/A1, an antiapoptotic member of theBCL2family, as well as an increased incidence of t(14;18); (2) “BCR/proliferation” which was defined by increased expression of BCR signaling cascade members (CD19, CD79a, BLK, SYK, PLC2, MAP4K), transcription factors such as PAX5, OBF-1, E2A,BCL6, STAT6, MYC, and cell cycle regulatory genes including CDK2 and MCM with an association withBCL6translocation; and lastly, (3) Host Response (“HR”) which was enriched for genes involved in the tumor microenvironment including T-cell mediated immune responses and classical component pathway[14].Unlike other classification systems, the three clusters had similar 5-year OS (53%, 60%, and 54% [P=0.53] for OxPhos, BCR/Proliferation, and HR, respectively), suggesting that this hierarchy would be a more useful tool for novel therapeutic development rather than for prognostication.To this effect, a comparison of this clustering system vs.COO classification demonstrated that the two systems described different aspects of DLBCL biology.Nearly half of the tumors classified as BCR/proliferation cluster (53%) and OxPhos (46%) were classified as GCB, while the remaining tumors in each classification were either ABC-or ‘unclassifiable’.On the other hand, the majority of HR cluster tumors were designated ‘unclassifiable’based on COO designation.
While extremely informative and prognostic, GEP is rarely used in the real world setting due to several limitations, including the ability to rapidly produce these results in an informative way and, most importantly, the reproducibility of these classifications at the community level.As such, other methods have been developed using immunohistochemistry (IHC), including Hanset al.[15], Choiet al.[16]and Tallyet al.[17]algorithms in an attempt to rapidly replicate GEP classifications with an additional benefit of significantly lower cost.The most frequently used IHC method, Hans’ algorithm, proposes utilizing CD10, BCL-6 and MUM-1 expression to differentiate between GCB vs.Non-GCB DLBCLs[15], whereas Choi uses GCET1,MUM1, CD10, BCL6 and FOXP1[16]in order to do so.The Tally method uses a similar antibody panel as Choi with the notable difference that antibody expression is not reviewed in a sequential manner but denoted by a score of 0-2, reserving the evaluation of LMO2 if an equal number of GCBvs.ABC genes/score are present[17].Interestingly, these three IHC methods incorporate CD10, echoing earlier studies in the 1990s supporting the use of CD10 to broadly differentiate DLBCL subtypes[7-9].Although quite rapid,IHC methods are riddled with limitations including inter-user inconsistencies and datasets suggesting inaccurate classifications using IHC as compared to GEP.For instance, Gutiérrez-Garcia and colleagues discovered that approximately 30-50% of GCB-DLBCLs and 15-25% of ABC-DLBCLs were incorrectly classified by IHC[18].In a separate study, Hans’ algorithm failed to demonstrate a difference in OS between GCB-DLBCL and non-GCB DLBCL, whereas classification of subtypes using Lymph2Cx assay, a GEP platform comprising of a 20 gene panel that can be applied to FFPE tissue samples[19], was able to demonstrate both a 5-year OS and disease-free survival difference (96.6%vs.77.1%, 96.6%vs.79.2%,respectively) in patients with GCB-vs.ABC-DLBCLs[20].In fact, the Lymph2Cx assay misidentified 2%DLBCL tumor samples compared to assignments made by the gold standard GEP[21].To put this in context,the COO assignment assessed by Tally, Hans, and Choi IHC-methods led to a misassignment rate of 6%,9%, and 17%, respectively[15-17].Subsequently, the Lymph2x assay was validated in a large cohort of DLBCL patients (n= 335) treated with R-CHOP therapy and confirmed that COO was associated with clinical outcomes independent of MYC/BCL2 expression and IPI score[22].Thus, given its improved accuracy compared to IHC, faster turn-over compared to gold standard GEP and the ability to predict prognostic outcomes, Lymph2Cx was thought to be a more applicable diagnostic tool.Along those lines, the ROBUST clinical trial[23,24], a phase III clinical trial that investigated the merits of combining lenalidomide to R-CHOP in ABC-DLBCL patients, utilized the Lymph2Cx assay as a companion diagnostic in order to rapidly identify COO.Despite theoretically serving as a real-time GEP assay, the adaption of Lymph2Cx to the ROBUST study led to a delay in treatment initiation due to logistical hindrances such as central review of tumor specimens resulting in an inadvertent introduction of selection bias for patients with lower risk disease that ultimately may not have benefitted from the addition of lenalidomide to R-CHOP[24,25].Therefore, although less robust, IHC categorization is still in universal use, with GEP assays often reserved for clinical trial studies or academic institution applications.
Novel classifications beyond cell of origin
In the era of precision medicine, on-going attempts to better target specific mutations and aberrations have led to additional sub-classifications that extend beyond COO.GEP, next-generation sequencing and copy number variation evaluations have made this possible, permitting an increasingly detailed understanding of DLBCL genomic profiles.
In an integrative analysis utilizing whole-exome sequencing and transcriptome sequencing of 1001 newlydiagnosed DLBCL patients, Reddyet al.[26]identified 150 genetic drivers of the disease and, in turn,characterized the functional impact of these genes using an unbiasedin vitroCRISPR screen[26].CRISPR screening of a panel of DLBCL cell lines led to the identification of 35 oncogenes, with 9 genes identified in a subtype-specific pattern:EBF1,IRF4,CARD11,MYD88andIKBKBwere essential in ABC-DLBCL lymphomagenesis, whereasZBTB7A, XPO1, TGFBR2andPTPN6were critical for the survival of GCB-DLBCL.With this, a genomic risk model incorporating 150 driver genes, COO designation, and MYC/BCL2 expression was able to better stratify response to therapy compared to the IPI clinical model, as well as models relying only on COO, MYC and/or BCL2 expression.
In another attempt to better understand the genetic landscape of DLBCL, Schmitzet al.[2]characterized 574 DLBCL samples (NCI cohort) utilizing exome and transcriptome sequencing, array-based DNA copy number analysis and targeted amplicon resequencing.Four genetic subtypes were elucidated: (1) MCD defined byMYD88 L265PandCD79Bmutations, (2) N1 based onNOTCH1mutations, (3) BN2 based onBCL6fusions andNOTCH2mutations; and (4) EZB characterized byEZH2mutations andBCL2translocations.Stratification of outcomes based on this molecular classification revealed that the MCD and N1 subtypes had inferior OS compared to the BN2 and EZB counterparts after standard of care R-CHOP therapy.The LymphGen algorithm builds upon the aforementioned 4-class taxonomy, identifying 7 subtypes: MCD, N1, BN2, ST2 (characterized bySGK1andTET2mutated), A53 (defined by aneuploidy withTP53inactivation), and divides the EZB category into two dichotomous subtypes: EZB MYC-positive vs.EZB MYC-negative[4]based on the presence or absence of a Double Hit signature (discussed below).This genetic algorithm validated the inferior OS observed in the MCD and N1 tumor subtypes.Ultimately, the LymphGen algorithm was able to successfully classify more patient tumors in the NCI cohort (63.1%)compared to the initial 4 class method (Schmitzet al.[2], 46.6%).Thus, it is clear that not all tumors fall discretely into these categories, indicating that there is still a need to build upon these current molecular programs.
In the cluster (C) classification system, five genetically unique subgroups were defined, with a sixth cluster(C0) consisting of a small number of tumor samples (12 of 304) with no common defining drivers[3].In this system, C3 (defined byBCL2, KTM2D, CREBBP, EZH2, PTENmutations) and C5 (based onMYDL265P,CD79B, ETV6, PIM1, GRHPR, TBL1XR1, BTG1mutations andBCL2gains) exhibited markedly inferior progression-free survival (PFS) compared to C1, C2, and C4.Interestingly, C3 and C5 are comprised mostly of GC-DLBCL and ABC-DLBCL cases, respectively, reinforcing the notion that there is more to classifying tumors based solely on COO.Together, this data suggests that the heterogeneous landscape of DLBCL is more complex than the COO classification, and that these new molecular classifications may lend themselves to precision-based medicine approaches.
Double hit lymphoma signature
High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements, termed Double Hit(DHL) or Triple Hit lymphomas (THL) have dismal outcomes after standard chemoimmunotherapy[27-29].These diagnoses rely on FISH analysis to evaluate and confirm rearrangements of MYC, BCL2 and/or BCL6.In order to define the genetic landscape of DHL/THL, Ennishi and colleagues analyzed 157 GCBDLBCL primary tumor samples, of which 25 were DHL/THL based on FISH, and developed a DHL signature (DHITsig), comprising of 104 differentially expressed genes[5].Twenty-seven percent of GCBDLBCLs were characterized as positive for the DHITsig, with only 50% of these tumors harboringMYCandBCL2translocations.Traditionally, GCB-DLBCLs are thought to originate from the light zone of the GC based on GEP; however, tumors that were DHITsig positive displayed more similarities to B-lymphocytes from the intermediate zone of the GC, suggesting a completely unique COO for DHL/THL.This was recapitulated by the presence of intermediate zone genes in the DHITsig.The prognostic implication of the DHITsig was validated in two separate cohorts (BC Cancer Center Cohort,n= 261; and Reddyet al.[26]cohort,n= 511), both of which demonstrated clear survival disadvantage for patients with the DHITsig,which paralleled the outcomes of ABC-DLBCL patients.Interestingly, along withMYCandBCL2mutations,TP53, EZH2, CREBBP, DDX3XandKMT2Dwere more frequently mutated in the DHITsig positive tumors as compared to the DHITsig negative tumors, suggesting possible therapeutic interventionsfor the treatment of DHITsig DLBCLs.In an attempt to create a more applicable tool for routine use, the authors condensed the 104-gene panel into 30 genes of essential importance, which was then added to the Lymph2Cx assay, and referred to as DLBCL90.The DLBCL90 was able to distinguish and prognosticate between DHITsig positive GCB-DLBCLs, DHITsig negative GCB-DLBCLs, ABC-DLBCLs and unclassifiable tumors.The DHITsig provides further support for the adaptation of complex genetic evaluation to help identify true poor-risk patients.
To further elaborate upon the DHITsig and classification of GCB-DLBCL, Song and colleagues combined results obtained from IHC, GEP, DLBCL90, FISH analysis for DHL/THL, copy number analysis, and targeted deep sequencing of 334 genes, and established four distinct groups within the broad GCB category:GCB1 defined as DHITsig positive with TP53 inactivation; GCB2 characterized by DHITsig positivity;GCB3 being DHITsig negative and EZB mutated and/or BCL2 translocation (EZB-like); and lastly, GCB4 which were both negative for DHITsig and EZB-like characteristics[6].Using 87 primary patient samples,patients assigned to the GCB2 and GCB4 categories displayed superior prognosis after R-CHOP therapy compared to the GCB1 and GCB3.This survival advantage was validated in an additional 188 GCB-DLBCL cases.Along these lines, the majority of cases fell under the GCB4 group (51%), perhaps, driving our older observation that GCB-DLBCLs, as a whole, have favorable outcomes after standard R-CHOP.However, it is clear from this study and others that not all GCB-DLBCLs (and ABC-DLBCLs) behave the same,supporting the need to validate a universal assay that can quickly output these novel molecular subclassification designations.
Limitations of novel genetic classifications
Although these novel classification systems and assays add to our understanding of DLBCL and have prognostic implications, the question still persists on how applicable and readily available these assays are to everyday use.We have been able to apply some of these diagnostics to clinical trial studies, however, we have yet to evaluate these new hierarchies in a prospective manner.Ultimately, the true implication of these novel clusters or sub-classifications requires prospective validation in parallel with the evaluation of novel therapeutics applied in a precision medicine fashion.Moreover, these assays often rely upon central laboratories, which illustrates another limitation - the ability to replicate these diagnostic tests from one laboratory to another with ease and efficiency, all the while maintaining the accuracy of the assay.Until we are able to quickly, and without compromising accuracy, replicate these assays in local or commercialized laboratories, the pervasive use of these diagnostic tools will be limited to clinical trial and academic applications.Furthermore, as we continue to probe into the genetic background of DLBCL, we are likely to find even smaller subsets based on unique genetic and mutational characteristics, which in turn will impact clinical trial design.Although these classifications are not yet ready for universal use, the wealth of information obtained from these analyses has revealed potential targets for novel drug development and will greatly assist in our attempts to improve the clinical outcomes of DLBCL patients.
Potential of circulating tumor DNA in the management of Diffuse Large B-cell Lymphoma
Liquid tumor biopsies have been developed in both solid and hematological malignancies as both a predictive and diagnostic tool that obviates the need for potentially invasive biopsies, and is currently an emerging tool for the DLBCL management.Circulating cell-free DNA (cfDNA) are DNA fragments that are routinely released into circulation by apoptotic or necrotic cells.In general, cfDNA are cleared from the plasma by the renal system, liver and/or spleen as well as nucleases[30].Circulating tumor DNA (ctDNA)specifically refers to cfDNA fragments that originate from tumor cells and thus far has been investigated in diagnostic, therapeutic and prognostic scenarios.Polymerase chain reaction (PCR)-based methods that identify single point mutations, such asMYD88 L265Pin primary central nervous system lymphoma[31,32],were one of the earliest iterations by which ctDNA was evaluated in lymphoma patients.This method isrelatively low cost and efficient but has the limitation of analyzing only select mutations with only a few mutations occurring with a high enough frequency to make PCR broadly applicable to disease subtypes.
As such, next-generation-sequencing (NSG) has revolutionized our ability to evaluate a panel of relevant mutations in a lymphoma-subtype or cancer-specific way.In order to achieve a broader applicability in lymphoma, the detection of clonotypic immunoglobulin (Ig) rearrangements through high throughput sequencing (IgHTS) was developed and used in early minimal residual disease (MRD) and relapse detection studies[33,34].However, IgHTS is limited by the amount of total cfDNA present in plasma and cannot provide information regarding mutational profiling or genetic drivers.Unlike IgHTS, the cancer personalized profiling by deep sequencing (CAPP-seq) is able to target many genetic aberrations with a sensitivity of 2.5 part per 105cfDNA[35]and is able to be diagnostic and therapeutic by providing mutational profiling.In fact,as compared to IgHTS, CAPP-seq has a higher sensitivity in both tumor and plasma ctDNA detection, and is able to provide COO information, as well as detect DLBCL relapse earlier than IgHTS[36].
Several studies have evaluated baseline ctDNA and correlated levels to overall tumor burden[33,34], as well as clinical outcomes with patients harboring > 2.5 log he/mL of ctDNA at baseline having inferior event free survival (EFS) at 24 months in both the front-line and salvage settings[37].Moreover, the role of interim ctDNA monitoring in the upfront and salvage setting revealed two excellent responder groups: the early molecular response (EMR: 2 log decrease after 1 cycle of treatment) and major molecular response (MMR:2.5 log decrease after 2 cycles of therapy)[37].Treatment naive patients who achieved an EMR or MMR had superior outcomes as measured by EFS and OS at 24 months compared to patients who did not achieve these landmarks (EMRP= 0.0015, andP< 0.001; MMR,P< 0.001 andP= 0.047, respectively).In the patients receiving salvage therapy, EMR was also prognostic and associated with improved EFS (EFS 100%vs.13%,P= 0.011).
In order to improve the depth and sensitivity for ctDNA MRD, Kurtz and colleagues developed phased variant enrichment and detection by sequencing (PhasED-seq) which identifies multiple clustered mutations on a single DNA molecule.These mutations are present on the same strand of DNA and occur in predicted areas of the genome, driven by aberrant somatic hypermutation by activation-induced deaminase[38].In fact, PhasED-seq was able to outperform CAPP-seq in the ability to detect MRD after 2 cycles of therapy as well as at end of treatment.
Ultimately, ctDNA is a promising and emerging biomarker in lymphoma, including DLBCL, that can overcome potential limitations of tissue biopsies and radiographic scans all the while providing molecular/tumor profiling information, risk stratification and disease/surveillance monitoring.The bar for sensitivity of ctDNA detection continues to rise, with the newest method of PhasED-seq improving the depth of detection compared to prior NSG methods.Although ctDNA seems logistically an appropriate biomarker and diagnostic tool to further develop, it inherently does not provide information regarding the interaction of the tumor and tumor microenvironment and has its limitations with low variant allele frequencies.Ultimately, prospective studies are needed to validate the exact role of ctDNA for MRD assessment, the development of possible risk-adapted trials (de-escalation or escalation of treatment), and lastly, as clinical surveillance following the completion of treatment.
EVOLVING TREATMENT OPTIONS
Standard of care
Despite extensive work that has been accomplished in understanding and elucidating the diverse pathological drivers of DLBCL, several attempts at improving our first-line therapy for DLBCL have failed.
Although several rational attempts have been made to improve the R-CHOP backbone[23,39-49], with a particular emphasis on ABC/non-GC-DLBCL subtype, only two trials have been successful in achieving their primary endpoints over the past two decades.For instance, clinical trials evaluating the merit of adding lenalidomide to R-CHOP backbone(E1412 and ROBUST)[24,50]as well as ibrutinib (PHOENIX)[49], all seemed like promising interventions, but only one (E1412) of the three trials successfully demonstrated an improvement in PFS[50].Although there is a multitude of reasons why these trials differ in the outcome,leading thoughts include different methods and approaches to identifying ABC/non-GC DLBCL patients,the longer interval from the time of diagnosis to initiation of treatment in the ROBUST study compared to E1412, and lastly, differing dosing of lenalidomide in ROBUST vs.E1412.It is not until recently, with the addition of a novel antibody-drug conjugate against CD79, polatuzumab-vedotin, to the R-CHOP backbone[51], that we have observed an improvement in the first-line setting in a large randomized, phase III clinical trial setting.Other attempts to improve upon the R-CHOP backbone are currently on-going including the addition of other monoclonal antibodies, such as tafasitamab and bispecific antibodies, all of which can be applied irrespective to COO, similar to polatuzumab-vedotin, thus circumventing the prerequisite need to identify DLBCL genetic subgroups prior to enrollment/treatment.
For those who relapse, the standard of care remains chemoimmunotherapy salvage therapy followed by consolidation with autologous stem cell transplant (ASCT)[52,53], at least for those who are deemed clinically fit.However, given that DLBCL is often a disease of the elderly, ASCT may not be a feasible option, leaving a clear unmet need for these patients.The more recent approvals in CAR-T cellular therapy seem promising[54-57], especially for patients who are chemo-refractory to R-CHOP and/or salvage chemotherapy;however, it is clear that the majority of patients will relapse after CAR-T cell therapy.In the following section, we will discuss recently FDA-approved agents and their applicability to the treatment paradigm of DLBCL as well as novel agents in the pipeline that leverage our understanding of the various oncogenic drivers elucidated by the recent molecular classifications [Figure 1], [Table 2].For discussion purposes, we have organized the following section based on COO, rather than the new classification systems, and highlight some potential agents that can target high-risk subgroups as delineated by LymphGen and cluster taxonomies.
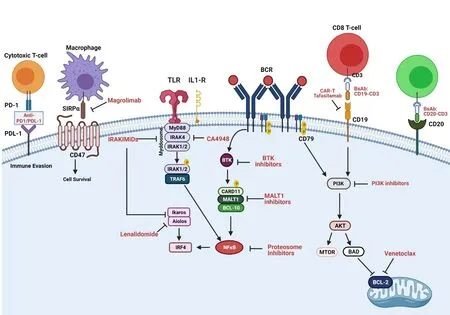
Figure 1.Novel Agents Targeting Specific Biological Pathways in Diffuse Large B-cell Lymphoma.B-cell receptor (BCR) pathway is affected in both GC- and ABC-DLBCL subtypes.Chronic BCR signaling, as manifested by BCR clustering, is a hallmark of ABC-DLBCL,and mimics antigen-dependent BCR activation.Tonic BCR signaling (antigen-independent, no BCR clustering) is a characteristic of GCDLBCL, and is NF-κB-independent.Mutations in proteins involved in BCR signaling and interrelated pathways include CD79B, CARD11,A20, and LYN.MYD88 L265P mutations lead to lymphomagenesis through activation of the TLR and IL1R pathways independent of ligand stimulation and have been highlighted in newer molecular classifications.Small molecule inhibitors (IRAK4, BTK, MALT1, PI3K,BCL2), protein degraders (IRAK4), antibody/antibody-conjugates (Polatuzumab-vedotin, Tafasitamab, magrolimab), and cellular therapy (CAR-T) have been developed in order to target these specific aberrations to mitigate lymphomagenesis.BCL2: B-cell lymphoma 2; BCR: B-cell receptor; BsAb: Bispecific antibodies; BTK: Bruton tyrosine kinase; ILR: Interleukin receptor; MALT1: mucosaassociated lymphoid tissue lymphoma translocation protein; PI3K: phosphoinositide 3-kinase; SIRPα: signal-regulatory protein alpha.Created with BioRender.com.

Table 2.Novel therapeutics and combinations for diffuse large B-cell lymphoma
Targeting specific molecular drivers
Novel agents for the treatment of ABC-DLBCL
ABC-DLBCLs are characterized by the overactivation of B-cell receptor (BCR) and NFκB pathways.Loss of function mutations/homozygous deletions in A20[58,59], and gain of function mutations in CARD11[58,60],CD79 A/B[61], MYD88[62], as well as the role of CARD11-BCL1-MALT1 complex[63-65], have all been implicated as drivers of the NFB pathway in ABC-DLBCLs, and attempts to target these pathological drivers have been made in the past several years.Based on the new molecular classifications, BCR-dependent NFκB signaling is prevalent in the MCD, A53, and BN2 subtypes, with MCD DLBCLs having an intermediatepoor prognosis after standard of care[4].
One of the first approaches of targeting NFB in ABC-DLBCL was the interrogation of proteasome inhibitors, such as bortezomib.Although thought to have pleotrophic effects, bortezomib and other proteasome inhibitors have been used to downregulate NFB activity via inhibition of IB degradation.However, two frontline studies comparing the addition of bortezomib to R-CHOPvs.R-CHOP in ABCDLBCL patients failed to reach their primary endpoints of improved PFS[47,48].
Another more selective approach to target the ABC-subtype is the inhibition of the Bruton tyrosine kinase(BTK), which is an essential member of the BCR pathway.Ibrutinib, a first-in-class covalent inhibitor ofBTK, demonstrated single-agent activity in patients with R/R DLBCL with an ORR of 37% (14/38) in ABCDLBCL patients, and a more impressive response in patients with dual mutations in MYD88 and CD79A/B,albeit a small sample size (ORR 80%, 4/5)[66].Phelanet al.[64]elucidated a possible mechanism driving ibrutinib responses in ABC-DLBCL, describing a My-T-BCR complex comprising MYD88-TLR9-BCR that interacts with mTOR within endolysosomes to activate downstream NFkB signaling.The My-T-BCR complex was found to be present in ABC-DLBCL cell lines as well as primary patient tumors that responded to ibrutinib, and responses were also noted in the absence of dual mutations in MYD88 and CD79A/B.Given these promising pre-clinical and clinical data, a phase III randomized clinical trial was launched in patients with non-GC DLBCL but failed to demonstrate any additive benefit to R-CHOP[49].In a preplanned analysis, patients younger than 60 years of age benefited based on an improved PFS and event-free survival (EFS); on the other hand, those older than 60 years of age suffered from increased toxicity leading to reduced intensity therapy, likely contributing to reduced efficacy.One can also postulate that using a less robust strategy of IHC to identify high-risk patients as non-GC DLBCL may have also impacted the results.Nonetheless, retrospective GEP analyses of available tumor samples from the PHOENIX trial were also analyzed, and there was no improvement of EFS, PFS or OS when adding ibrutinib to R-CHOP.Nextgeneration BTK inhibitors are in the investigation for the treatment of DLBCL, including the less toxic acalabrutinib.In the relapsed/refractory (R/R) setting, acalabrutinib has been evaluated as a single agent(ORR of 24%, 5/21)[67]and more recently in a 4-arm Phase I study (PRISM) that evaluates acalabrutinib in combination with one of the following: AZD9150, an antisense oligonucleotide against STAT3 mRNA;AZD6738, an inhibitor of ATR; magrolimab (anti-CD47) and rituximab; or AZD5153, a BRD4 inhibitor[68].Acalabrutinib is also being studied in the upfront setting (NCT0400294, NCT03571308) in combination with R-CHOP or DA-R-EPOCH.Unlike ibrutinib, early data suggests that acalabrutinib does not compromise the ability to administer R-CHOP regardless of age[69,70].

Ab: Antibody; BR: Bendamustine+Rituximab; BsAb: bispecific antibody; CR: complete response; DLBCL: diffuse large B-cell lymphoma; FL:follicular lymphoma; ITT: intent to treat; PR: partial response; MMAE: monomethyl auristatin E; NHL: non-Hodgkin lymphoma; PROTAC:proteolysis targeting chimera; PI3K: phosphoinositide 3-kinase (PI3K) inhibitor; R-CHP: Rituximab, Cyclophosphamide, Doxorubicin, Prednisone;R-CHOP: Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, Prednisone; R/R: relapsed/refractory.
IRAK4 has been a recent target for the treatment of MYD88-mutated DLBCLs, which represent a subgroup with inferior outcomes (MCD and C5 subgroups).IRAKs are key mediators of the toll-like receptor (TLR)and interleukin-1 receptor (ILR) signaling pathways and are essential in the innate immune system,inflammation, apoptosis and cancer biology.TLR/ILR signaling is mediated through recruitment of MYD88, an adaptor molecule, which forms the core of the Myddosome complex along with IRAK4[71,72].Upon ligand binding, TLR/ILR dimerize or oligomerize, leading to the recruitment MYD88 which then interfaces with IRAK4 through death domains[71].The most common MYD88 mutation substitutes leucine 265 for a proline (L265P) and leads to constitutive activation and formation of the Myddosome (MYD88-IRAK4-IRAK1/2), independent of ligand binding, leading to activation of downstream effectors including NF-κB transcriptional pathway and thus oncogenesis[73].Given its role in DLBCL lymphomagenesis, IRAK4 has been identified as a potential therapeutic target.The IRAK4 inhibitor, CA-4948, has completed phase I evaluation in R/R NHL, including DLBCL, with an identified recommended phase II dosing (RP2D) of 300mg oral twice daily[74].The treatment was overall well tolerated, with no grade 5 adverse events, and only two treatment associated discontinuations secondary to increase amylase and rash.At therapeutic treatmentdoses ( 200mg BID), a range of tumor reduction was observed (5-67%).Twenty of the 31 patients enrolled had available information for MYD88 status, and of the 20, two harbored anMYD88mutation, with one patient achieving a partial response.Based on a synergistic relationship observedin vivo, the combination of ibrutinib in conjunction with CA-4948 is currently being evaluated in a multicentered, phase I study in patients with R/R NHL[74,75].
Another novel method of targeting IRAK4, has been the development of Proteolysis Targeting Chimeras against IRAK4.These small molecules target specific proteins for degradation by cross-linking pathogenic proteins to E3 ubiquitin ligases, leading to ubiquitination of the target protein.This serves as a rationale mechanistic approach to eliminate oncogenic signaling.These first-in-class heterobifunctional degraders represent an innovative approach that could revolutionize cancer therapy.In contrast to IRAK4 kinase inhibitors, a strategy of targeting IRAK4 degradation would completely nullify the protein’s dual role as both a kinase and scaffolding protein, and potentially circumnavigate internal resistance.First-in-class heterobifunctional selective IRAK4 degrader, KYM-001[76], demonstrated potent efficacy in MYD88-mutated lymphomas.IRAKIMiDs, which are novel IRAK4 degraders that utilize a cereblon binder leading to dual targeting of IRAK4 degradation and capitalizing on immunomodulatory(IMiD) biology, also demonstrates activity in MYD88 mutant DLBCL[77].In pre-clinical models, IRAKIMiDs in conjunction with ibrutinib, venetoclax and umbralisib have proven to be synergistic[78], suggesting a possible multidrug platform using IRAKIMiDs as a backbone in order to potentially downregulate several oncogenic pathways simultaneously.Further clinical studies are required to validate these promising pre-clinical data with plans for KT-413, the lead IRAKIMiD, to enter clinical evaluation in 2022.
The development of MALT1 inhibitors is being intensively investigated and has demonstrated pre-clinical activity in ABC-DLBCL[79-86].BCL10 gain of function mutations, most commonly found in BN2/C1 subgroups[2,3], have been associated with increased MALT1 and NF-κB activity, as well as resistance to ibrutinib[87].When exposed to JNJ-67856633, a MALT1 inhibitor, ibrutinib-resistant ABC-DLBCL cell lines were characterized by growth inhibition, supporting the therapeutic application of MALT1 inhibitors in ABC-DLBCL, especially those with BCL10 mutation and ibrutinib-resistance.At this time, a phase I study(NCT03900598) is currently on-going investigating JNJ-67856633 in R/R NHL (NCT03900598)[88].
PD-L1 expression has been associated with inferior outcomes and found in higher expression in ABCDLBCL as compared to GCB-DLBCL[89,90], and therefore has been another target for the advancement of DLBCL treatment.It is thought that tumor cells exploit immune checkpoint pathways in order to evade the host immune system.However, single-agent immune checkpoint inhibition (nivolumab) displayed limited efficacy in R/R DLBCL, demonstrating an ORR of 3% and 10% for patients who relapsed after ASCT and were ineligible for ASCT, respectively[91].Notably, the incidence of 9p24.1 alterations was low (16% low-level copy gain, 3% amplification) and may partially explain the poor efficacy results after administration of nivolumab.The addition of PD1/PD-L1 inhibitors in the upfront setting has also been evaluated,postulating that upfront utilization may lead to better outcomes by optimizing a relatively more intact immune system.As such, pembrolizumab (anti-PD1) in combination with R-CHOP produced an ORR of 90% (27/30) and a PFS of 83% (NCT02541565)[92].A Phase I/II clinical trial investigating the addition of durvalumab (anti-PD-L1 antibody) to R-CHOP or R2CHOP [lenalidomide + R-CHOP backbone](NCT03003520) was initiated, however the R2-CHOP + durvalamub enrollment (Arm B) was halted due to increased deaths in patients with Multiple Myeloma treated with durvalumab and lenalidomide[93,94].The CR rate at the end of induction was 54% and 67% for Arm A (n= 43) and Arm B (n= 3), respectively, with consolidation therapy with durvalumab offered to patients who responded.Another potential platform to incorporate immune checkpoint inhibition is the notion of sequential therapy, during which PD1/PD-L1inhibition is used as induction, followed by R-CHOP, and concluding with PD1/PD-L1 inhibition maintenance.The rationale behind this sequential therapy is to maximize the potential efficacy of PD1/PDL1 inhibition by avoiding corticosteroid-induced immunosuppression that could be observed when adding immune checkpoint inhibition directly to the R-CHOP backbone.This is the approach that Hawkes and colleagues adopted, where avelumab is combined with rituximab as induction, followed by R-CHOP x 6 cycles, and avelumab maintenance x 6 cycles[95].Induction with avelumab and rituximab x 2 cycles led to a CR rate of 21% and ORR of 60%, followed by an ORR of 89% after six cycles of R-CHOP.Further studies will be needed to fully elucidate the role of immune checkpoint inhibitors in the frontline setting for DLBCL.
Targeting GC-DLBCL
Although patients with GCB-DLBCL generally have superior clinical outcomes compared to ABC-subtype,approximately 20% of GCB patients will relapse following standard of care R-CHOP.Moreover, our understanding of the molecular heterogeneity of DLBCL has revealed that DLBCLs withMYC-aberrations tend to have an inferior prognosis compared to those who do not harborMYC-derangements.Approximately 5% to 14% of DLBCL cases have been reported to carryMYCtranslocations[28,96], and of those, 58% to 83% will also be characterized by a second or third translocation targetingBCL2and/orBCL6,which are referred to as DHL or THL, respectively[97-99].In fact, DHL/THL patients have a median OS rate of less than two years[27,100-102].Moreover, MYC overexpression has also been associated with worse outcomes after first-line R-CHOP[97], and can also be frequently observed in conjunction with BCL2 overexpression(Double Expressors), which often coincide with an ABC-DLBCL categorization[27,29,103].Thus, it has been clearly established that R-CHOP therapy is suboptimal for the treatment of MYC-altered DLBCLs, and the current treatment paradigm favors more dose-intense approaches, such as DA-EPOCH-R, (Dose-Adjusted-Etoposide, Prednisone, Oncovorin, Cyclophophamide, Doxorubicin, Rituximab) in the upfront setting.
Pre-clinical data has demonstrated that the epigenome of GCB-DLBCL is dysregulated.Deregulation of histone methyltransferases, such as EZH2 and mixed-lineage leukemia (MLL), have been associated with the development of lymphomas, particularly GC-derived lymphomas[104-107].Given the recognition of EZH2 activating mutation in GC-derived lymphomas, such as follicular lymphoma (FL) and DLBCL, EZH2 inhibitors have been developed based on single-agent activity observed in pre-clinical models of DLBCL[104,108-119].Tazemetostat, a first-in-human EZH2 inhibitor, has been FDA approved for the treatment of R/R FL irrespective of mutational status.Despite initial promising responses observed in phase I investigation, a dedicated phase II clinical trial of tazemetostat in conjunction with prednisolone in DLBCL(NCT01897571) was closed after an interim assessment[120,121].Clinical investigation of tazemetostat in combination with R-CHOP is on-going at this time (EPI-RCHOP, NCT02889523).The addition of tazemetostat thus far does not seem to compromise R-CHOP dose-intensity[122].Valemetostat, a dual inhibitor of both EZH1 and EZH2, is postulated to have improved tumor suppression capabilities, and has demonstrated an ORR of 53% (n= 15) in patients with R/R NHL (B and T-cell lymphoma)[123].These EZH2 inhibitors would be ideal targets for patients in the EZB MYC-negative and C3 subtypes.
Heterozygous loss of function mutations in histone acetyltransferases (HATs) such asCREBBPandEP300are found in approximately 39% of DLBCL and FL[124]and cooperate with BCL6 to promote lymphomagenesis.The presence of CREBBP and EP300 haploinsufficiency has been associated with response to histone deacetylase (HDAC) inhibitors inin vitroevaluations[125]; however, it is unclear if this completely translates into the clinic.Nevertheless, single-agent HDAC inhibition for the treatment of DLBCL has modest activity with a reported ORR of 17.1% after treatment with panobinostat[126].Fimepinostat, a first-in-class dual HDAC and phosphoinositide 3-kinase (PI3K) inhibitor, demonstrated anORR of 55% (5/9) in patients with DLBCL[127], leading to a dose expansion trial in R/R DLBCL.In the expanded component of this trial, 14 patients with MYC-altered DLBCL, defined as MYC translocated or amplification with ≥3 copies in >20% of cells, were included with an observed ORR of 64% and median duration of response (DOR) of 13.6 months[128].In a pooled analysis of phase I and phase II study of fimepinostat, a total of 105 patients with R/R DLBCL were enrolled, of which 60 patients had MYC-dysregulated DLBCL (classified as the presence of MYC translocation of MYC overexpression ≥ 40%).The ORR was noted to be 23.3% (14/60), with a median time to response being 2.5 months[129].Given these promising results in MYC-deregulated DLBCL, a phase I trial investigating the merits of fimepinostat in conjunction with rituximab or venetoclax, or both (NCT01742988) is underway.Given the presence of recurrent epigenetic dysregulation in GCB-DLBCL, pre-clinical and clinical investigation of potential synergistic drug combinations involving multiple epigenetic agents and/or other small molecule therapeutics is warranted.
Bromodomain and Extra-terminal (BET) proteins have also been linked to the activation of MYC and BCL2 signaling pathways, making it a rationale target for DHL[130].Pre-clinical studies have demonstrated that treatment with JQ-1, an early BET inhibitor, and venetoclax, a BCL-2 inhibitor along with standard vincristine or doxorubicin, led to significant growth delay of DHL/THL cells compared to single-agent treatment[131].Moreover, exposure to CPI203, a BET inhibitor, can overcome acquired resistance to venetoclax in pre-clinical DHL models[132], supporting the possible applicability of BET inhibitors for the treatment of DLBCL, particularly DHL/THL.Although theoretically promising, several BET inhibitors have entered clinical investigation, however, none to date have gained FDA approval.
The application of venetoclax for the treatment of DLBCL addresses chemotherapy resistance secondary to BCL-2 overexpression, especially in patients with double expressor and DHL[133].Single-agent activity of venetoclax in R/R DLBCL is modest, with a reported ORR of 18%[134].Recent studies have incorporated venetoclax into the backbones of R-CHOP (CAVALLI study)[135]and DA-EPOCH-R (ALLIANCE 51701 trial)[136], specifically targeting the DHL/Double Expresser populations.In the R/R setting, venetoclax is being explored in various combinations including ibrutinib/rituximab (NCT03136497), RICE(NCT03064867), Selinexor (NCT03955783), and ibrutinib/prednisone/obinutuzumab/revlimid(NCT03223610, ViPOR).Ultimately the role of venetoclax in the treatment of DLBCL is yet to be firmly established.
Therapeutics agents with activity irrespective of COO
Despite our advances in understanding the molecular heterogeneity of DLBCL, one can argue that perhaps the fastest and smoothest approach to the advancement of DLBCL treatment would be to develop novel drugs that can be applied across subtypes without the need for detailed molecular testing.This, of course,would be a shiftawayfrom a precision medicine approach, a central theme in cancer treatment, but in the real-world setting may be more applicable as it would overcome the hindrances and delays that often come with the requirement of specific and sensitive assays.A primary example of this success would be rituximab[137].Along those lines, novel antibody and antibody-drug conjugates have been developed for lymphoid malignancies, including three recent FDA approvals in the R/R setting.Polatuzumab vedotin, an antibody-drug conjugate against CD79b linked to monomethyl auristatin E, has been approved in conjunction with rituximab and bendamustine (BR) after two failed therapies for transplant-ineligible patients.A phase Ib/II study demonstrated both a superior CR rate (40%vs.17.5%,P= 0.026) and median OS (12.4vs.4.7 months; HR:0.42; 95%CI: 0.24-0.75,P=0.002) of polatuzumab-BR compared to BR alone[138].Polatuzumab-vedotin has also been evaluated in combination with R-CHP (vincristine omitted due to overlapping toxicities) in a randomized phase III trial for treatment-naive patients[51], resulting in a lowerrisk of progression/relapse/death as compared to standard of care R-CHOP (HR: 0.73; 95%CI: 0.57-0.95;P=0.02).However, there was no difference in CRs (78%vs.74%, respectively,P= 0.16) or OS (2-year: 88.7%vs.88.6%).Polatuzumab-vedotin is currently under investigation in combination with salvage chemotherapy regimens such as R-GemOx and R-ICE, as well as in combination with venetoclax/rituximab[139], with the latter demonstrating a relative safe toxicity profile and median PFS and OS of 4.4 months and 11.0 months,respectively.
Tafasitamab, a humanized anti-CD19 monoclonal antibody, gained FDA approval in combination with lenalidomide for R/R DLBCL (> 1 prior line of therapy) after the L-MIND study demonstrated an ORR and CR rate of 54% and 32%, respectively, with a median PFS of 16.2 months[140].In an updated analysis, the median DOR was reported to be 34.6 months[141].The FIRST-MIND study is evaluating the addition of tafasitamab plus lenalidomide to R-CHOP in intermediate-high risk DLBCL (NCT04134936)[142].Similarly,loncastuximab tesirine, an antibody-drug conjugate against CD19, received FDA approval in 2021 after results demonstrated an ORR of 48.3%, with a CR rate of 24% (35/145) and a PR rate of 24% (35/145) in a heavily pretreated R/R DLBCL population[143].
CD47 is expressed by malignant cells as a means to evade macrophages and other members of the innate immune system[144].The novel anti-CD47 monoclonal antibody, magrolimab, is currently in development for R/R DLBCL as well as acute myeloid leukemia.This monoclonal antibody effectively inhibits a “do not eat me” signal and induces phagocytosis of lymphoma cells by inhibiting the interaction of CD47 and SIRP[145].Thus far, a phase Ib study of magrolimab in conjunction with rituximab for the treatment of R/R DLBCL or FL demonstrated an objective response and CR of 40% and 30%, respectively, for patients with DLBCL[146].A notable adverse event was anemia (attributed to the elimination of aging red blood cells) and infusion-related reactions which were circumvented by priming (1mg/kg) and subsequent maintenance dosing of magrolimab.
Bispecific antibodies (BsAb) are recombinant bispecific proteins that have the dual binding capacity,recognizing two different antigens, and in turn, are able to redirect T-cells to malignant cells in order to mediate a T-cell response and T-cell directed enhanced tumor cell death.Blinatumomab, the first-in-class CD3-CD19 BsAb, has been approved for relapsed acute lymphoblastic leukemia, with limitations of a short half-life requiring a continuous infusion.Next generation BsAbs have shown promising efficacy in R/R DLBCL, including relapse after CAR T-cell therapy, and are easier to administer compared to blinatumomab.Other benefits of BsAbs are that they are ‘off-the-shelf’ products, requiring no additional time for manufacturing, such as in the case of autologous CAR-T-cell therapies.Mosunetuzumab, a CD3-CD20 BsAb, is currently being studied in indolent and aggressive NHL and has demonstrated a single agent ORR of 37% (46/124) and CR of 19% (24/124) in patients with aggressive NHL[147].Notably, of the 30 patients who progressed after CAR T-cell therapy, 18 patients were available for response, with four patients(23%, 4/18) achieving a CR, and a total ORR of 39% (7/18).Twenty-eight percent of patients experienced cytokine release syndrome (CRS), with 1.4% of patients experiencing Grade 3 CRS, all of which resolved with the administration of tocilizumab.Neurological toxicity was observed in 44% of patients, with the majority being Grade 1 and 2 (27.4% and 12.6%, respectively).Mosunetuzumab is currently being evaluated in a host of clinical trials for NHL, including the GO40515 study (NCT03677141), in which mosunetuzumab is combined with CHOP or CHP-Polatuzumab in the first-line setting for DLBCL as well as a single agent in the treatment-naive elderly/unfit patients (NCT03677154)[148].
Several other CD3-CD20 BsAbs have entered clinical development as well.The phase I study of odronexatamab was conducted in a highly refractory patient population, with 80.3% determined to berefractory to their last line of therapy[149].In patients with R/R DLBCL who were CAR T-cell naive and treated at doses ≥ 80 mg (n= 10), the CR and ORR rate was 60% with a duration of response (DOR) of 10.3 months.Those who were refractory to prior CAR-T cell therapy and treated at ≥80 mg (n= 21), remarkably had an ORR of 33.3%, CR rate of 23.8% and median DOR of 2.8 months.The rates of Grade 3 CRS and neurotoxicity were 6.3% and 2.3%, with one observed Grade 4 CRS event noted.Epicoritamab, a subcutaneous administered BsAb against CD3-CD20, with proposed improved side effect profile due to its route of administration, has completed phase I evaluation with RP2D of 48mg based on pharmacokinetic profiling[150].In DLBCL patients treated at ≥ 48 mg (n= 7), all patients achieved a response, with 2 of 7(28.6%) attaining a CR and 5 of 7 (71.4%) achieving a PR.Four patients were treated previously with CAR-T cell therapy, all of which achieved a response (2 CRs, 2 PRs).Lastly, glofitamab, a 2:1 BsAb that bivalently binds to CD20 malignant B-cells and monovalent binding to CD3 on T-cells, has demonstrated superior potency in pre-clinical models compared to 1:1 BsAbs in conjunction with pre-treatment with obinutuzumab (used to mitigate CRS)[151].The phase I/Ib evaluation of glofitamab demonstrated an ORR of 50.0% in 24 evaluable aggressive NHL patients, and a CR rate of 29.2%[152].The most common AEs were CRS(57.9%), none of which were Grade 3 or higher, pyrexia (31.6%), neutropenia (28.9%), thrombocytopenia(28.9%) and hypophosphatemia (28.9%).Ultimately, the postulated enhanced tumor killing of this newer class of 2:1 CD20-CD3 BsAb compared to traditional 1:1 binding will require further clinical trial investigation.
Another emerging agent is the trispecific antibody CMG1A46 which engages CD19-CD20-CD3 simultaneously, targeting not only CD19+/CD20+ DLBCL, but also those patients who relapse and lose expression of CD20[153].In vivoandin vitrostudies have demonstrated improved efficacy compared to 1:1 BsAbs, and a phase I clinical trial is planned for the near future.
Another unique class of therapy that leverages immunological aspects is Chimeric antigen receptor T-cell therapy (CAR-T cell therapy).Thus far, autologous CAR-T cell constructs against CD19 (axicabtagene ciloleucel, lisocabtagene maraleucel, tisagenlecleucel) have been FDA approved in patients with R/R DLBCL who have failed two or more lines of therapy.Three large phase III clinical trials have attempted to move the CAR-T cell indication to the second-line setting, particularly evaluating patients who are deemed chemo-resistant (primary refractory or relapsed ≤ 12 months from first-line chemoimmunotherapy).Two of the three randomized trials demonstrated clinical benefit compared to autologous stem cell transplant(Zuma-7, TRANSFORM)[154,155], while the third randomized trial failed to meet its primary endpoint(BELINDA)[156].Several possibilities have been postulated to reconcile these differences in outcomes,including the high proportion of patients enrolled in the BELINDA trial experiencing progression of disease at week 6 (prior to CAR-T cell infusion) in part due to a delay in CAR-T cell manufacturing (median time to infusion: 52 days, range 31-135 days).
Additionally, as more follow-up data has become available, it has become apparent that approximately 30-50% of patients who initially respond to anti-CD19 CAR-T cell will suffer a relapse, with the majority occurring within one year of infusion[157].The mechanisms behind some of these relapses have been attributed to limited persistence of the cellular products and downregulation/loss of CD19, leading to antigen escape[158].Given these findings, potential combinations with targeted agents in combination with autologous anti-CD19 CAR T-cell have been studied.Qinet al.[159]demonstrated that the combination of ibrutinib with lisocabtagene maraleucel led to improved CAR T-cell effector function translating into improved survival of CD19 tumor-bearing mice[159].Lenalidomide has demonstratedin vivosynergy with both anti-CD19 and anti-CD20 CAR-T cell products[160], and more recently has been documented to induce responses in patients relapsing post-CAR T-cell infusion likely due to both an anti-tumoral effect andimmunomodulatory mechanisms[161].Another way to leverage the immune axis is the addition of PD-1/PDL-1 inhibitors to CAR-T cell products as the expression of PD-1 is enhanced after receipt of anti-CD19 CAR-T cell infusion[162,163].As proof-of-principle, a case report of a patient with refractory DLBCL who progressed rapidly on ZUMA-1, was administered nivolumab 3 mg/kg (11 days after infusion with axicabtagene ciloleucel), leading to a rapid re-expansion of CAR T-cell product within 48 hours of receipt of nivolumab and eventual reduction of tumor volume after completing two additional nivolumab doses[164].This, along with pre-clinical data, has laid the foundation for ZUMA-6, which is evaluating axicabtagene ciloleucel in combination with atezolizumab (anti-PD-L1 antibody) in patients with refractory DLBCL[165].There is also emerging dating supporting the use of PI3K inhibitors during CAR T-cell production as it can enhance the expansion of CD8+ CAR T-cells, with durable persistence and increased efficacy[166].In addition to combination therapies, novel CAR-T cell products have been explored in order to overcome the resistance associated with anti-CD19 CAR T-cell therapy, including novel autologous CAR-T cell products against CD20[167]and CD22[168], tandem CAR-T cell therapies in which CD19/CD20 are engaged simultaneously[169,170], and the development of allogenic CAR-T cell therapy, with the latter obviating the need for leukapheresis.
Although not immunologically-based, selinexor, an oral inhibitor of XPO1-mediated nuclear transport, has also recently been FDA approved, and its therapeutic index is observed across subtypes.Selinexor demonstrated an ORR of 28% (36/127) with 12% achieving a CR (15/127) in the R/R DLBCL population,and a DOR of 9.3 months (95%CI: 4.8-23.0)[171].The most common grade 3-4 AEs were thrombocytopenia (n= 58), neutropenia (n= 31), anemia (n= 28), fatigue (n= 14), hyponatremia (n= 10), and nausea (n= 8).Subgroup analysis demonstrated that responses were similar irrespective of COO, age, gender, previous therapy, refractory status and history of ASCT.
FUTURE DIRECTIONS: WHAT THE FUTURE OF DIFFUSE LARGE B-CELL LYMPHOMA CARE MAY LOOK LIKE
Despite our deeper understanding of the complex molecular landscape of DLBCL as well as several attempts to leverage this understanding in a logical matter, we have only recently improved upon our standard of care frontline chemoimmunotherapy.Although R-CHOP has remained a cornerstone in therapy for two decades, several improvements in the R/R setting have been observed, ranging from targeted oral molecular agents to antibody-drug conjugates to cellular therapy.In the past five years, the only drugs/therapies that have successfully achieved FDA approval for DLBCL (CD19 CAR T-cell therapy, polatuzumab vedotin,tafasitamab, loncastuximab, selinexor) are not used in a subtype-specific matter.In fact, these newer molecular DLBCL classifications are not readily available in the community and will require the development of rapid, reproducible assays in order to be widely adapted.A focus on the integration of immunologic and small molecule agents that are universal to all DLBCL subtypes into our treatment paradigms avoids the need to develop highly specific and costly molecular assays to identify a target population.Along those lines, until rapid and reliable assays are developed, one rational way to improve the R-CHOP backbone is the addition of agnostic agents such as monoclonal antibodies that avoid the need for detailed molecular subtyping that often leads to a longer diagnosis-to-treatment time, while reserving a more precision based approach in the R/R setting.Once the development of a reliable assay is created based on companion studies in the R/R clinical trial setting, the assay can be applied in the frontline setting, where we have come to acknowledge the importance of the diagnosis-to-treatment interval[25].Alternatively, in order to avoid delay in the initiation of treatment and permit deep molecular analysis, another option is to incorporate pre-phase therapy as performed by Pfreundschuh and colleagues[172]in the upfront or even R/R clinical trial setting.There is also a dire need to develop broader eligibility criteria to evaluate novel precision medicine-based agents and their associated companion diagnostics as it is clear that high-riskpatients with baseline organ dysfunction have inferior EFS as well as higher lymphoma-related mortality and often are not able to enroll into classically designed clinical trials[173].Ultimately, the future may be a mixture of these elements: a new platform that combines novel immunological agents in conjunction with specific small molecule agents tailored to a certain subtype of DLBCL.Regardless of the route, given the complexity of these new DLBCL subtypes, it is likely that the future of clinical trial investigation will require combinational therapy in order to target multiple oncogenic pathways to optimize therapeutic benefit and avoid the development of drug resistance.
CONCLUSIONS
The therapeutic platform for the treatment of DLBCL has been inundated with novel agents spanning from small molecule inhibitors to bispecific antibodies to cellular therapy.Although numerous agents have been introduced, the majority have not impacted frontline therapy favorably.The use of drugs that are agnostic to COO or DLBCL subtype may circumvent the permissive use of these complex genetic analyses in order to prevent delays in treatment initiation.Future investigations should focus upon synergistic drug combinations that are tailored for specific DLBCL molecular subtypes, all the while optimizing genetics assays in order to do so.The development of rapid, reliable, and affordable companion genetic assays that can allow us to classify patients into these newer DLBCL subclassifications is a necessity and will allow us to translate clinical trial findings into real-world use.At the same time, we must leverage the potential of ctDNA and other translational tools for diagnostic, prognostic and therapeutic applications.Nevertheless,in parallel with our greater understanding of the genetic heterogeneity of DLBCL comes a plethora of opportunities to develop novel agents that target underlying driver pathological pathways, and ultimately,the future of DLBCL treatment paradigms will continue to evolve, including chemotherapy-free approaches.
DECLARATIONS
Author’s contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation: Jennifer K.Lue, Grzegorz S.Nowakowski
Availability of data and materials
Not Applicable.
Financial support and sponsorship
None.
Conflicts of interest
JKL received research support and advisory board service from Kymera Therapeutics; advisory board service from Astex Pharmaceuticals, consultancy for Daiichi Sankyo and Kura Oncology; and Speakers’Bureau from AstraZeneca.GSN:Grant research Support:Celgene/BMS, MorphoSys, Roche, Nonostrings;Consultant: Celgene/BMS, Denovo Biopharma, Kite Pharma, Kymera Therapeutics, MorphoSys, Roche;Other: Ryvu Therapeutics (scientific advisory board)
Ethical approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Copyright
© The Author(s) 2022.
 Journal of Cancer Metastasis and Treatment2022年3期
Journal of Cancer Metastasis and Treatment2022年3期
- Journal of Cancer Metastasis and Treatment的其它文章
- Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice
- The bone marrow niche landscape: a journey through aging, extrinsic and intrinsic stressors in the haemopoietic milieu
- The WHO 2021 thymoma classification: a work in progress
- Chronic activation of MUC1-C in wound repair promotes progression to cancer stem cells
- AUTHOR INSTRUCTIONS