
三个中国无脉络膜症家系的临床与遗传分析*
2022-05-05 02:02李鹏程秦亚运何晓婕唐朝晖刘木根
华中科技大学学报(医学版) 2022年2期
李鹏程, 秦亚运, 何晓婕, 唐朝晖, 刘 飞, 刘木根△
1华中科技大学同济医学院附属协和医院眼科,武汉 430022 2华中科技大学生命科学与技术学院,武汉 430074 3华中科技大学医院,武汉430074
无脉络膜症(choroideremia,CHM;OMIM:#303100)是一种少见的X连锁的遗传性视网膜疾病,其患病率约为1∶50000~1∶100000,以感光细胞、视网膜色素上皮(RPE)和脉络膜的进行性退化为主要特征[1-3]。CHM基因(OMIM:*300390)是无脉络膜症的唯一致病基因,编码REP-1蛋白[4]。REP-1蛋白对于Rab蛋白的异戊二烯化修饰和正确定位是必需的,其功能障碍会导致细胞囊泡运输异常和蛋白质错误定位[5-6]。CHM的主要症状为进行性加重的夜盲、渐进性视野缩小,晚期逐渐出现中心视力下降,与视网膜色素变性(retinitis pigmentosa,RP)的临床症状高度重叠,极易被误诊为RP[7]。因此,遗传分析对于CHM的精准诊断十分必要。本文对3个中国无脉络膜症家系进行了临床和遗传学研究,现将结果报道如下。
1 资料与方法
1.1 参与者与临床检查
本研究招募了2016~2021年在华中科技大学同济医学院附属协和医院和华中科技大学医院就诊的3个中国汉族非近亲家系。纳入标准:①满足无脉络膜症的临床诊断标准,有夜盲病史,特征性眼底表现及家族史符合X连锁遗传特征;②通过分子遗传学方法明确CHM基因突变。排除有其他眼部疾病及不能配合完成眼科检查者[8]。所有参与者或其法定监护人均已签署知情同意书。本研究经过华中科技大学伦理委员会的审查与批准。所有参与者在协和医院进行了全面眼科检查,包括最佳矫正视力测量(best corrected visual acuity,BCVA)、裂隙灯观察、眼底彩照和光学相干断层扫描(spectral domain optical coherence tomography,SD-OCT)等。采集参与者的外周血标本2 mL,采用试剂盒(TIANamp Blood DNA Kit)提取基因组DNA,并测定浓度和纯度[9-10]。
1.2 全外显子组测序及生物信息学分析
全外显子组测序和生物信息学分析由药明康德公司(WuXi AppTec)完成。外显子捕获和建库测序采用SureSelect_Human_Exon_V5试剂盒(Agilent Technologies,Santa Clara,USA)和HiSeq 2500测序平台(Illumina,San Diego,USA)。根据生信分析结果和美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南,挑选候选基因和突变进行致病性评级[11]。家系3患者就诊时携带外院进行的二代测序报告。
1.3 基因突变的验证
首先,通过Sanger测序确认全外显子组测序得到的候选突变是否存在。其次,采用限制性片段长度多态性分析(restriction fragment length polymorphism,RFLP)对家系内成员进行共分离验证,并验证候选突变是否存在于200例正常人对照中。CHM基因的c.186C>G突变会破坏SexAⅠ的酶切位点。通过一对引物(F:5′-TTGGGATCTGAGAGGCCTACT-3′,R:5′-CGCACCCGAGCTC-TATTTATAT-3′)扩增包含突变位点的CHM基因片段,再用限制性内切酶SexAⅠ(New England BioLabs)对PCR产物进行酶切(37℃,4 h),DNA凝胶电泳观察条带大小变化。
2 结果
2.1 家系图与一般特征
本研究招募的3个家系的成员关系见图1。家系1纳入3名男性患者。患者Ⅱ-1,10岁左右出现夜盲症状,约20岁时在外院接受抗青光眼手术治疗,来院就诊时有严重白内障且拒绝手术治疗,无法进行眼底检查,故本研究排除该患者。家系1中Ⅱ-3、Ⅱ-5的父母均无夜盲或视力下降症状,而Ⅲ-4的父母也表现正常,提示遗传方式可能为X连锁隐性遗传(图1A)。家系2、3各纳入1名第3代男性患者;而家系2、3的患者Ⅱ-1均有夜盲症状和视力障碍,初步判定为患者,但在外院就诊,未进行详细的眼科检查,故未纳入本研究。家系2、3遗传方式与家系1类似(图1B、1C)。三家系中均无近亲结婚史。纳入的5例患者初诊年龄22~49岁,初诊中位数年龄39岁。

A:家系1;B:家系2;C:家系3;'□:正常男性;○:正常女性;■:男性患者;⊙:女性携带者;↑:先证者图1 X连锁视网膜变性家系图谱Fig.1 Maps of X-linked retinal degeneration families
2.2 患者临床表现
2.2.1 视力 所有5例患者均从10岁左右开始出现夜盲,部分患者逐渐出现中心视力下降。5例10眼均接受了BCVA检查。患者BCVA介于0.02~0.60之间(小数视力),其中80%患眼视力≥0.5,明显好于之前关于国人报道中的平均视力0.28[8]。值得注意的是,家系1的Ⅱ-3、Ⅱ-5患者初诊时均存在晶体后极部局限性皮质浑浊,未接受手术治疗。家系1的3名患者随访5年视力均无明显变化。家系2的1名患者于2021年复诊,随访半年,视力无明显变化。家系3患者双眼视力0.6。
2.2.2 眼底改变 家系1先证者Ⅱ-5眼底检查显示双眼视盘边界清晰,视网膜色素上皮及脉络膜毛细血管层广泛萎缩、消失,导致脉络膜大血管层和巩膜暴露,中周部视网膜有骨细胞样色素沉积(图2A),双眼黄斑中心凹反光消失。患者Ⅱ-3的左眼黄斑灰白色隆起,右眼与先证者类似;患者Ⅲ-4的眼底情况好于先证者(图2A)。女性携带者Ⅰ-2未见明显眼底异常(图略),Ⅱ-8见广泛椒盐状脱色素改变(图2A)。
家系2患者眼底所见与上述患者基本一致;双眼萎缩性改变及色素沉积波及黄斑区,较家系1患者程度更严重;且其右眼黄斑大片视网膜下白色脉络膜新生血管(CNV)性增殖膜(图2B)。家系3患者眼底见双眼萎缩性及色素沉积波及黄斑区,巩膜暴露更明显,余与上述患者类似(图2C)。家系1和家系2的4名患者于2021年复诊,眼底均无明显变化。

A:家系1中患者Ⅱ-5和Ⅲ-4眼底中周部可见暴露的大血管和巩膜以及片状色素沉积,携带者Ⅱ-8周边视网膜可见大量的椒盐状脱色素改变;B:家系2先证者Ⅲ-2眼底可见严重的视网膜色素上皮和脉络膜萎缩、大量片状色素沉积以及右眼黄斑区视网膜下脉络膜新生血管性增殖膜;C:家系3先证者Ⅲ-2眼底可见严重的视网膜色素上皮和脉络膜萎缩,色素沉积及巩膜暴露;OD:右眼;OS:左眼图2 彩色眼底照片Fig.2 Color fundus photographs
2.2.3 SD-OCT下的形态改变 家系1先证者Ⅱ-5的SD-OCT结果显示右眼黄斑保留相对完整的视网膜层次和结构,但外丛状层与外核层间出现微小的黄斑囊样病变(cystoid macular lesion,CM)及外层视网膜管状结构(outer retinal tubulation,ORT);外核层在黄斑区保留的情况相对好于周边区域;黄斑中心凹以外的区域可以观察到突然丢失的外层视网膜以及脉络膜显著变薄和对应处巩膜反射信号的增强(图3A)。患者Ⅱ-3右眼中心视网膜厚度约为295 μm,左眼可见严重的大面积椭圆形全层视网膜囊样病变(横径约12 mm,竖径约6 mm,中心视网膜厚度约1000 μm)(图3B)。患者Ⅲ-4的SD-OCT表现与先证者右眼类似。先证者母亲Ⅰ-2和妹妹Ⅱ-8双眼黄斑结构正常(图略)。

A:家系1先证者Ⅱ-5右眼黄斑区SD-OCT扫描图,黄斑区见囊样改变(白色箭头),中心凹外见外层视网膜管状结构(红色箭头),黄斑中心凹以外可以观察到突然丢失的外层视网膜以及脉络膜显著变薄和对应处巩膜反射信号的增强(黄色箭头);B:家系1患者Ⅱ-3左眼黄斑区SD-OCT扫描图,左眼出现大面积波及全层视网膜的囊样病变;C:家系2先证者Ⅲ-2双眼SD-OCT图,右眼见视网膜下CNV性高反射团;OD:右眼;OS:左眼图3 患者的SD-OCT表现Fig.3 SD-OCT presentation of the patients
家系2先证者Ⅲ-2的SD-OCT结果显示右眼黄斑中心凹区域存在明显视网膜下CNV性增殖膜,左眼黄斑结构相对完整,双眼萎缩性改变与家系1患者改变类似(图3C)。家系3患者双眼见萎缩性改变与上述患者类似(图略)。
所有10只眼,8只(80%)可见不同程度黄斑囊样病变,其中7眼为微小病变。7眼(70%)可见ORT改变,且大部分位于黄斑中心凹以外伴有RPE萎缩的区域。患者临床资料汇总见表1。
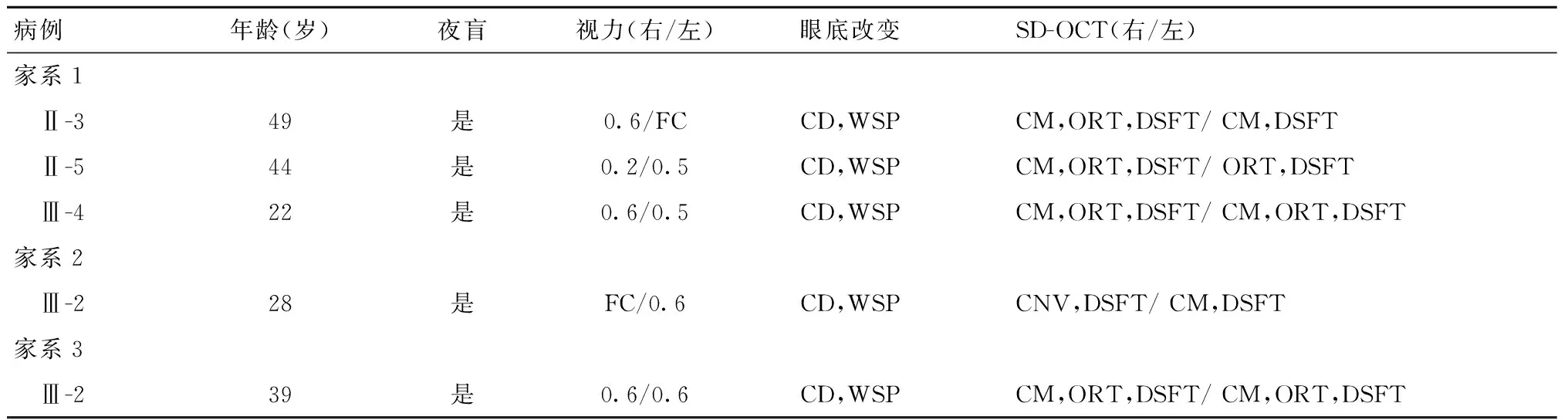
表1 患者临床资料Table 1 The clinical data of patients
2.3 致病基因和突变鉴定
3个家系均不存在近亲婚史,且都存在父母视力正常而子女发病的情况。患者均为男性,而其女性同胞(姐妹)均不发病。结合家系图和患者的临床表现,我们对3个家系的初步诊断为X连锁隐性遗传的视网膜色素变性或无脉络膜症。
对于家系1,在排除X连锁RP致病基因RPGR和RP2后,我们对先证者Ⅱ-5进行了全外显子组测序和生物信息学分析,共发现6个位于X染色体上的候选突变(CHM c.186C>G;AKAP4 c.2191G>C;TSC22D3 c.472C>T;SHROOM2 c.4541A>C;ARMCX6 c.131G>A;ARMCX6 c.333T>A)。经过Sanger测序验证(图4A)和家系内共分离分析(图4B),最后确定CHM为该家系致病基因。CHM基因的c.186C>G突变是一个未见报道的无义突变,可导致CHM基因第62位酪氨酸密码子TAC转变为终止密码子TAG,产生一个含61 aa的截短蛋白(p.Tyr62*)。家系中有夜盲症状者均为男性且携带该突变,先证者母亲Ⅰ-2和妹妹Ⅱ-8为该突变的携带者(图4B)。
对于家系2,我们直接对CHM基因进行了Sanger测序,结果发现了一个CHM基因的c.189+1G>A突变(图4C)。该突变是一个剪接位点突变,曾经在一个西班牙家系中报道过[12],可导致3号外显子跳跃,产生一个含44 aa的截短蛋白,具有明确的致病性。

A:家系1先证者Ⅱ-5存在c.186C>G突变(黑色下划线);B:CHM基因突变在家系1中的共分离分析,正常人为2个较小条带,患者只有1条未切开条带,携带者则为3个条带;C:家系2先证者Ⅲ-2存在c.189+1G>A剪接位点突变(红色箭头)图4 CHM基因突变位点验证Fig.4 Verification of CHM gene mutation site
家系3患者就诊时已进行了全外显子组测序。通过查阅测序报告,发现了一个CHM基因的c.808 C>T突变。该突变曾在一个中国人家系中报道过[7],为无义突变,可导致翻译在第270位密码子处提前终止,具有明确的致病性。根据遗传学分析结果3个家系最终均确诊为无脉络膜症。
3 讨论
无脉络膜症是一种X染色体连锁的遗传性脉络膜视网膜退化疾病,主要症状包括夜盲、渐进性视野收缩、视力下降和畏光。由于视网膜色素上皮层、外层视网膜、脉络膜血管层的渐进性萎缩,患者眼底暴露出白色巩膜和深层脉络膜血管[13]。由于发病率低,国内罕有大宗病例研究。韩筱煦等[8]报道了一项包含10例患者的研究,提示中国CHM患者的平均视力为0.28,35岁以上患者70%眼的视力低于0.5,且病情进展更快。与上述研究不同的是,本研究纳入的患者视力大部分大于0.5,即使在有白内障存在的情况下也明显好于此前国人的报道,并且3例患者随防5年视力无明显恶化。国外的一些大宗病例报道,如Heon等[14]基于60例患者的研究发现,30岁以下患者的视力与正常人比较,差异无统计学意义;50岁以上患者的视力明显低于0.62(小数视力)。Freund等[15]基于128例患者的研究发现,40岁以下患者视力基本正常。本研究与国外的这些大宗病例报道结论较为一致,而与国人的报道有所不同。但由于本研究样本量过小,还需要进一步扩大样本量进行跟踪研究。
黄斑囊样病变是CHM常见的SD-OCT改变。本研究中CHM患者黄斑囊样病变的发生率为80%,大部分患眼该改变仅限于外层视网膜且程度较轻,仅1眼可见囊样改变波及节细胞层。此外,家系1患者中有1眼黄斑区存在大面积的严重囊样病变,是导致其视力低下的重要原因。这一现象此前在中国CHM患者中未见详细报道[7-8],但我们观察到的发生率与国外报道相近。如,Murro等[16]的一项包括42例患者的回顾性研究,发现38%的患者(34.7%患眼)有此改变。Genead等[17]的研究报道了一组16例患者,发现70%的患眼有不同程度的黄斑囊样病变。这一现象之前也在视网膜色素变性等遗传性视网膜变性类疾病中被报道,其具体发病机制还不明确,可能与血-视网膜屏障功能异常,视网膜色素上皮细胞的功能障碍等有关[18]。由于这一现象发生率很高,对患者进行定期复查一旦发现视网膜厚度增加,可考虑给予碳酸酐酶抑制剂治疗[18],有可能阻止患者视力进一步下降。
ORT指位于视网膜外层的、环以高反射信号边缘的、圆形或卵圆形低反射信号空腔。本研究纳入的5名患者10眼ORT的发生率为70%,其部位多位于黄斑中心凹以外伴有视网膜色素上皮萎缩的区域,与之前关于国人的研究结果类似[8]。Murro等[16]也曾在15/18(83.3%)患者中观察到此现象。ORT是Bietti结晶样视网膜变性、视网膜色素变性等变性类疾病晚期的标志[19-20],通常被认为是变性的光感受器在邻近视网膜色素上皮和胶质细胞的影响下发生的重排[20]。但是这一改变与患者视力、疾病进展等方面的关系还有待进一步研究。
本研究中家系2先证者右眼黄斑区视网膜色素上皮与脉络膜之间存在高反射团,结合病史考虑为继发脉络膜新生血管,是此眼视力低下的主要原因。有文献报道1例CHM患者发生CNV引起严重的视力下降,但是半年后发生自发消退,视力恢复到0.8[21]。也有报道,抗VEGF药物贝伐单抗可以有效治疗继发性CNV[22]。然而,本例患者就诊时CNV已经呈视网膜下疤痕样改变,无黄斑区出血或水肿等活动性病变,失去治疗价值。CNV也见于其他多种视网膜变性类疾病[21],是视力严重下降的重要原因。定期复查,及时发现和治疗这类并发症显得非常重要。
CHM是目前已知的X连锁隐性遗传无脉络膜症的唯一致病基因[13],目前已有300多个CHM基因突变被报道,包括错义、无义、剪接等突变类型(Human Gene Mutation Database,http://www.hgmd.cf.ac.uk/ac/index.php),其中以无义突变出现的频率最高。本研究中发现的CHM基因新突变(c.186C>G,p.Tyr62*)属于无义突变,但由于十分接近剪接位点,也可能引起pre-mRNA剪接的改变[23],突变导致REP-1蛋白的翻译在非常靠前的位置终止,无法产生有功能的蛋白。本研究家系2发现的剪接突变(c.189+1G>A,)曾见于报道,但在中国人群中是首次发现。家系3的突变为无义突变(c.808C>T),在中国人群已有报道。3个突变均会导致CHM蛋白的截短,从而无法行使正常功能,导致未修饰的Rab蛋白错误累积并影响细胞蛋白质的转运,最终引起感光细胞、视网膜色素上皮和脉络膜的进行性变性[6]。根据ACMG致病性评级指南,这3个突变均可确定为致病性突变。
综上所述,本文报道了3个中国无脉络膜症家系的临床特征和遗传病因。该研究拓宽了CHM基因的突变谱,提示黄斑萎缩性改变、严重的黄斑囊样病变或脉络膜新生血管是无脉络膜症患者中心视力下降的常见原因,为今后的遗传咨询和基因治疗奠定了基础。
猜你喜欢
现代医药卫生(2022年16期)2022-12-07
中华眼视光学与视觉科学杂志(2022年5期)2022-11-14
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
实用肝脏病杂志(2022年2期)2022-03-21
国际眼科杂志(2021年2期)2021-02-01
临床眼科杂志(2020年5期)2020-12-13
诊断学(理论与实践)(2020年1期)2020-04-28
水产科学(2020年2期)2020-03-20
