
费托合成钴基催化剂微观结构研究进展
2022-05-04 05:59卢文丽王俊刚孙德魁马中义陈从标李德宝
燃料化学学报 2022年4期
卢文丽 ,王俊刚 ,孙德魁 ,马中义 ,陈从标 ,侯 博 ,李德宝
(1.中国科学院山西煤炭化学研究所 煤转化国家重点实验室, 山西 太原 030001;2.中国科学院大学, 北京 100049)
费托(F-T)合成是以煤、天然气、页岩气和生物质等含碳资源为原料经合成气(CO+H2)在催化剂的作用下合成液体燃料或化学产品的工艺方法。费托合成反应是在1925年由德国的Fischer和Tropsch发现的,简称为F-T合成。目前,费托合成采用的催化剂主要包括铁(Fe)、钌(Ru)、钴(Co)和镍(Ni)等,由于钴基催化剂在费托合成中有反应活性好、水煤气变换反应活性低、长链烃选择性高、CO2选择性低、催化剂使用寿命长等优点,一直以来在工业上有着广泛的应用前景[1,2]。
催化剂的组成、结构,尤其是微观结构对FT合成反应的产物分布至关重要。其中,催化剂的组成一般包括活性组分、载体和助剂。研究已表明,在钴基F-T合成中起活性作用的是金属钴而不是氧化态的钴。明确活性中心后,随着纳米技术及其表征手段的不断发展,活性中心的调控逐渐成为可能。研究者们通过成熟的纳米晶合成技术,可控合成出各种形貌和结构的金属钴纳米晶活性中心。长期以来,为了提高催化剂的F-T反应活性、产物选择性以及稳定性,人们对钴基催化剂的制备过程、微观结构以及各个组分在F-T合成反应过程中的影响等方面做了大量的研究,这些工作对工业上指导设计高效的钴基费托合成催化剂具有重要的意义。
本文综述了钴基催化剂的尺寸效应、晶相、晶面、微观活性位点、微观活性吸附位以及吸附行为对催化反应的影响,讨论了微观活性位表征方法的研究进展,为设计高效钴基催化剂和深入了解相关反应机理提供了参考和支持。
1 催化剂微观活性位的结构
在真实催化反应条件下,由于纳米粒子表面形貌的不均匀性和尺寸不同,其表面的几何结构、电子结构和晶体结构会有所不同,从而引起催化剂表面上催化反应活性的不同。同时由于表面不同的活性吸附位点,会影响基元反应的路径,呈现出不同的催化性能。一直以来,在多相催化中,直接识别起作用的催化剂的活性位点、尤其是微观活性位点仍是钴基催化剂研究的核心问题。
结构敏感性研究是多相催化研究中非常重要的研究课题之一。催化反应分为两大类,即结构敏感反应和结构不敏感反应,由催化剂活性中心的结构和催化反应之间的关系决定。在多相催化中,许多重要的反应如合成氨反应、费托合成反应等都具有结构敏感性。结构敏感反应是指当催化剂活性组分固定后,催化剂的转化率随着催化剂结构的改变而变化的一类反应。由于F-T合成反应是结构敏感反应,钴基催化剂的费托反应活性和产物选择性明显依赖于金属钴结构[3]。下面将对钴基催化剂的纳米尺寸效应、晶相效应、晶面效应以及微观活性位结构研究现状进行阐述。
1.1 纳米尺寸效应
金属纳米颗粒的尺寸效应对负载型金属纳米材料的催化活性和选择性有重要影响。F-T反应作为结构敏感反应,长期以来,许多研究人员致力于探究实现高催化活性和选择性的最佳钴粒径。
1992年Iglesia等[4]的早期研究认为F-T合成的转换频率(TOF)对10−200 nm的钴颗粒尺寸不敏感。之后,许多学者研究了更小的钴颗粒的尺寸对负载型钴基催化剂本征活性的影响。大量的研究工作报道了钴基费托催化剂的尺寸效应。Bezemer等[5]把钴负载在碳纤维上来研究钴颗粒尺寸对费托反应性能的影响。他们发现,当钴颗粒的尺寸小于8 nm(3.5 MPa)时,催化剂有明显的尺寸效应,CO加氢的转换频率(TOF)会随着钴颗粒尺寸的增加而增加,大于8 nm时费托反应活性与钴颗粒尺寸无关(图1)。Xiong等[6]研究发现,对于直径在8−10 nm以上的钴基催化剂,TOF值是稳定的,而对于更小粒径的催化剂,TOF值急剧下降。同样,邱成武等[7]考察了Co粒子尺寸对F-T反应性能的影响。结果显示,随着Co粒子尺寸的增大,CO转化率降低,在钴粒子尺寸为8 nm时TOF表现出最大值,COads和C*在Co粒子表面吸附强度适中并且比例恰当,使得催化剂表现出较高的F-T活性和产物选择性。2020年Qi等[8]研究了由介孔二氧化硅(MCF-17)负载的四种不同尺寸(3.2、5.5、8.6和11 nm)的钴纳米颗粒的尺寸效应,当钴纳米颗粒直径为11 nm时,C5+的选择性最高,CH4的选择性最低,选择性与颗粒尺寸的关系与Bezemer等[5]的研究结论一致。
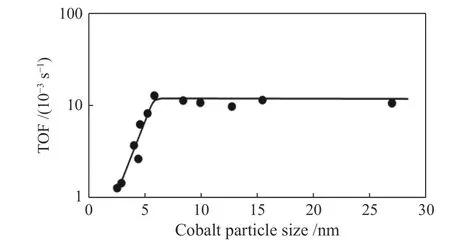
图1 钴颗粒尺寸对费托反应活性的影响[5]Figure 1 Influence of cobalt particle size on the TOF (220 ℃, H2/CO = 2, 0.1 MPa)[5]with permission from ACS Publications
目前,一些原位手段已经被用来研究费托合成中的尺寸效应。Den Breejen等[1]通过稳态同位素瞬态动力学分析(SSITKA)解释了小颗粒钴(小于6 nm)上催化活性低的原因。较小的Co粒子表面上低配位数的Co原子较多,CO在低配位数的Co上发生不可逆键合,部分边/角位点会被覆盖而留下了本征活性比较低的平台位点,从而导致催化活性低。Ralston等[9]采用时间分辨化学瞬态动力学方法(time-resolved chemical transient kinetics)研究了非稳态情况下4.3和9.5 nm Co表面的碳覆盖度,发现CO在9.5 nm Co表面解离的碳物种覆盖度是4.3 nm Co表面的两倍多。Tuxen等[10]利用原位软X射线吸收光谱表征手段发现,15 nm Co上的CO解离速率比4 nm Co上的CO解离速率快。
1.2 晶相效应
众所周知,在钴基催化剂费托反应过程中,金属钴是活性中心。钴基催化剂催化费托合成反应的结构敏感性,不仅在催化剂的尺寸效应上有所体现,同时也会反映在Co的晶相效应上,即不同晶相的金属Co显著影响着费托合成反应的活性和选择性。金属Co按原子堆积方式可以分为六方密堆(HCP)晶相和面心立方密堆(FCC)晶相,当HCP Co的含量较高时F-T合成催化活性较高,而当FCC Co含量较高时催化活性则相对较低。正如Mitchell等[11]发现,SiO2、Al2O3和TiO2载体上与H2还原干燥焙烧样品的活性相比,H2直接还原干燥样品的活性增加是由于HCP Co/FCC Co的物质的量比较高。
在钴基催化剂费托合成反应中,温度和钴基催化剂的颗粒尺寸大小都会影响HCP Co和FCC Co两种不同晶相的存在形式[12,13]。Tsakoumis等[14]采用还原-碳化-还原的方法来活化Re-Co/γ-Al2O3,当碳化后的还原温度为450 ℃时,HCP Co会发生部分相变转化为FCC Co。此外,Garces等[15]利用原位XRD方法研究了无载体的Co3O4在H2气氛中的还原过程,发现在 250−300 ℃时,Co3O4先被还原成CoO,再被还原成HCP Co,当还原温度高于400 ℃时只能得到FCC Co,但当进行分步还原时,在450 ℃下能够得到HCP Co和FCC Co的混合物。
大量的研究表明,HCP Co的F-T反应活性比FCC Co高。Liu等[3]基于第一性原理的动力学理论计算,研究发现,HCP Co比FCC Co具有更高的本征活性,其独特的晶体结构暴露出更多的活性位点。HCP Co倾向于直接解离C=O键,FCC Co倾向于通过氢助解离的方式进行(图2)。另外,不同的晶体结构材料可以显著影响催化剂反应活性和选择性。李微雪团队提出暴露特定高活性的HCP Co(10-11)晶面以提高活性位密度,实现高比质量活性、稳定的钴基催化剂的优化设计,从而提高催化性能。这些发现与van Santen等[16]的研究数据是相一致的,研究认为,CO的直接活化只有在台阶处的结构上提供足够低的活化能才能实现。
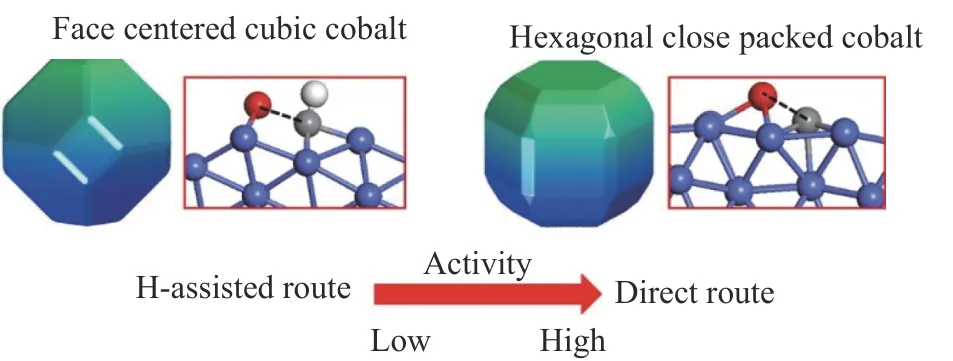
图2 FCC Co和HCP Co表面上C=O键直接活化和氢助活化[3]Figure 2 Breaking the C=O bond via the direct route and the H-assisted route on the HCP Co facets and FCC Co facets[3]with permission from ACS Publications
2018年Lyu等[17]成功采用了两步法合成了单一晶相的FCC Co和HCP Co,并且结合计算得到HCP Co的活化能垒比FCC Co低,真正意义上确认了HCP Co的高活性特征。采用XRD、SEM、TEM、TPR和H2-TPD等表征手段对钴基催化剂的理化性质进行了表征,证明了HCP Co的费托活性比FCC Co要高,对于两种晶相的钴基催化剂来说,HCP Co的活化能比FCC Co低约40 kJ/mol,前者的活化能要低得多(图3),这与计算研究结果一致。结果表明,HCP Co是费托合成中CO解离的首选相。
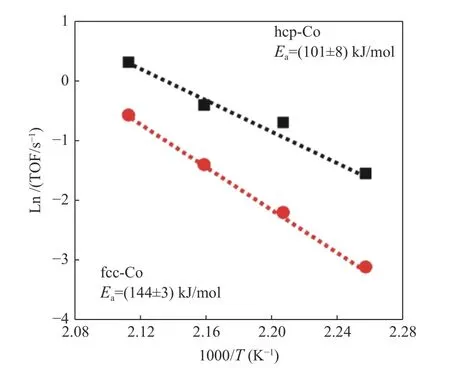
图3 不同Co基催化剂上CO的TOF阿伦尼乌斯图Figure 3 Arrhenius plot of CO turnover frequency over different Co catalysts
钴基催化剂的微观结构主要包括晶相结构、晶面结构以及微观活性位点结构,其中,微观活性位点又可分为边、角、棱、界面、甚至更加微观的B5、B7位点等。因此,可以利用钴的不同晶相结构来控制催化剂表面结构和形貌,从而能够提高催化剂的本征活性和活性位密度,对设计高活性催化剂具有重要意义。
1.3 晶面效应
了解和利用催化剂纳米材料的结构-性能关系是催化科学一个重要的问题,催化剂的不同晶面结构会影响费托反应活性以及产物的选择性。
Liu等[3]基于第一性原理的动力学理论计算,HCP Co上的四个晶面包括(11-21)、(10-11)、(10-12)和(11-20),均表现出比FCC Co最高活性的(100)晶面更高的CO 解离速率(图4)。
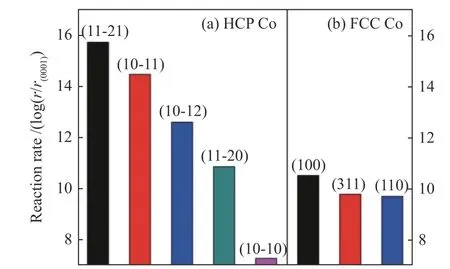
图4 HCP Co(a)和FCC Co(b)上低覆盖度下CO的解离速率[3]Figure 4 Calculated reaction rates r for CO dissociation on (a)HCP Co and (b) FCC Co at low coverage[3]with permission from ACS Publications
Zijlstra等[18]通过DFT理论计算发现,在高的CO覆盖度下,在Co(0001)表面,对CO解离来说,电子-电子排斥明显增加;在Co(11-21)表面,电子-电子之间的排斥作用并没有因为CO共吸附物的存在而增加。这种截然不同的趋势可以很好地解释为什么CO解离几乎不受台阶-边缘位置CO共吸附物的影响。Petersen等[19]也运用密度泛函理论对FCC Co(321)和FCC Co(221)在扭折和台阶位点上研究CO直接解离和氢助解离两种途径,结果表明,在低覆盖度下,CO直接解离是更具有优势的。同时, FCC Co 缺陷部位活性的增加并不会超过高活性 HCP Co(10-11)和(11-21)部位的活性(图5)。
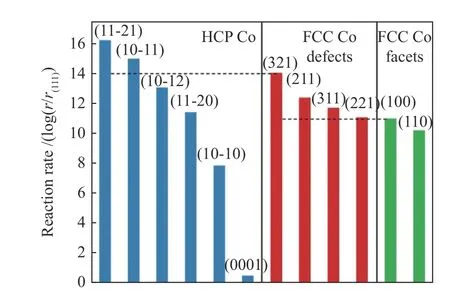
图5 在500 K下计算低覆盖度下CO在HCP Co(蓝色)、FCC Co(绿色)和FCC Co缺陷位点(红色)的解离速率已按FCC CO(111)计算的CO解离速率归一化[19]Figure 5 Calculated low-coverage CO dissociation rates at 500 K (in site–1 ·s–1) on HCP Co facets (blue), FCC Co facets(green), and FCC Co defect sites (red)Rates have been normalized to the CO dissociation rate calculated for FCC Co(111)[19]with permission from ACS Publications
另外,钴晶面对费托反应产物的选择性和碳链增长机理也有显著的影响。Zheng等[20]发现,经过还原−碳化−还原处理的催化剂Co(101)和Co(002)比还原处理催化剂Co(111)的烯烃选择性高。另外,大量的实验揭示了Co3C(101)对低碳烯烃的生成是有利的,甲烷选择性低,而Co2C(111)活性低,甲烷选择性高。Zhao等[21]通过理论计算发现,Co(0001)晶面倾向于以HCO插入的方式进行碳链的增长。Su等[22]通过DFT研究发现,在低CO覆盖度下,Co(0001)晶面和台阶式的Co通过CO插入机理来进行碳链增长,而在开放的Co(10-11)晶面则更倾向于以碳化物机理的方式进行链增长。进一步分析发现,产生这种差异的原因主要是最不“饱和”中间体(如C/CH物种)的吸附不同。近期,Zhang等[23]利用DFT计算研究了占据HCP晶相钴催化剂28%的Co(10-10)和35%的Co(10-11)晶面上遵循碳化物机理通过RCH2CH2与单体CH2的耦合形成R'CH2CH2(R'=H或者RCH2)实现碳链增长,R'CH2CH2加氢实现碳链的终止。因此,钴基催化剂表面结构对CHx的生成显示出结构敏感性。
本课题组[24]采用溶剂热法合成了具有特殊形貌且暴露不同晶面的Co3O4(> 15 nm)。在无载体和助剂存在的条件下,排除载体、助剂以及颗粒尺寸对F-T合成反应性能的影响,从而在实验上首次确认了F-T合成反应过程中钴的晶面效应。研究发现,由Co3O4(112)还原后得到Co(10-11)的晶面结构,该结构具有更多的B5位、倾向于以碳化物机理的方式进行链增长、易吸附C/CHx关键中间物种等特点,使其具有更高的F-T反应活性和长链烃选择性(图6)。本课题组的研究结果表明,可以通过选择性暴露Co3O4的特定晶面来提高FTS反应的活性和选择性,为合理设计高性能FTS催化剂开辟了途径。Lyu等[17]在实际的FTS反应条件下,也同样证明HCP Co(10-11)晶面表现出最高的FTS活性和C5+产物选择性,具有最低的表观活化能和CH4选择性。
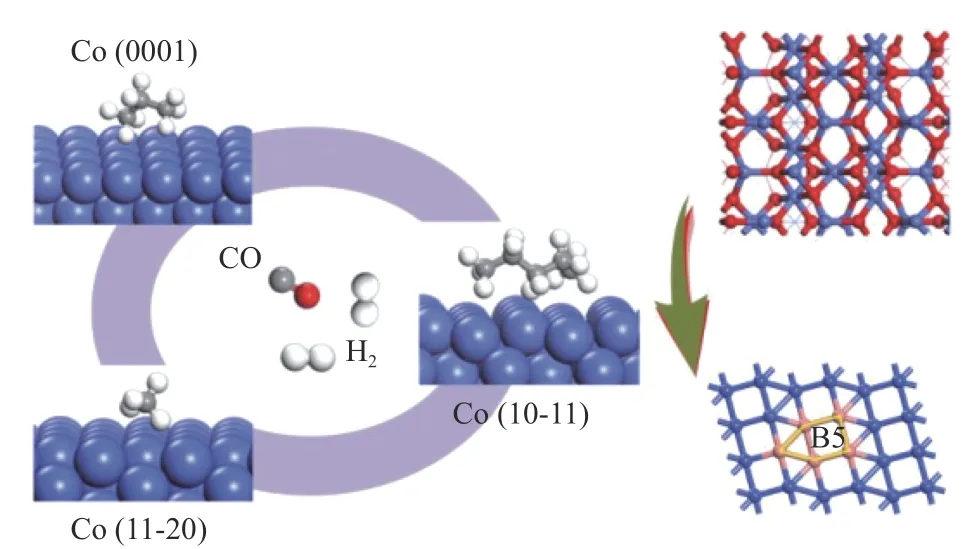
图6 Co(10-11)、 Co(0001)和 Co(11-20)表面的碳链增长能力示意图[24]Figure 6 Schematic diagram of the carbon chain growth ability for Co(10-11), Co(0001), and Co(11-20) surfaces[24]with permission from ACS Publications
1.4 微观活性位的类型
1.4.1 台阶、平面位点
众所周知,当金属纳米粒子小于10 nm时,其颗粒表面的角、边和台阶等会多过平台位置成为主要暴露的活性位点[25],即小颗粒尺寸的Co纳米颗粒拥有大量的低配位表面位点。
Agrawal等[26]应用分子动力学模拟钴纳米颗粒,以了解晶相对费托催化相关表面位点分布的影响,发现可能导致HCP Co纳米颗粒催化活性更高的原因是HCP Co纳米颗粒上平台和台阶/扭结位点的比值较高(10%−15%)。进一步,Boeller等[27]提出了一个现场原位扫描隧道显微镜研究,对Co(0001)型催化剂在高达950 mbar 的合成气压力和约500 K的温度下进行实验。数据表明,单原子台阶密度与在线测量的TOF有明显的相关性,表面台阶就是F-T合成反应中Co(0001)模型催化剂上的活性位点。Pestman等[28]研究了低压费托反应条件下的相关活性位点,关于暴露特定晶面的纳米粒子认为台阶位点负责链的增长,而平面位点则有助于甲烷的形成。相似地,Qin等[24]对有目的合成的暴露不同晶面的钴基催化剂进行了实验,发现暴露于台阶位点的褶皱晶面具有更高的费托反应活性和长链烃选择性,而暴露于平坦位点的晶面主要产生甲烷。Zijstra等[18]通过微动力学模拟,研究发现Co(11-21)的甲烷选择性较低,原因是在台阶-边缘位点C中间体的牢固结合抑制了甲烷化。
1.4.2 B5位点
CO在B5位点的解离会比在平坦的表面更为活跃,同时通过配位数(CN)和中心对称性(CS)这两个参数共同对B5位点进行分析和鉴定[29,30]。B5位通常由一个平面中的四个原子和另一个平面中的第五个原子形成。
Agrawal等[26]应用分子动力学模拟钴纳米颗粒,结果表明,HCP Co具有更高的本征活性,原因之一是HCP Co和FCC Co颗粒之间的B5位置分布明显不同。B5位的数目与配位数为10或11原子的存在密切相关。当配位数为11时,HCP Co纳米颗粒具有两个峰,分别对应于两种不同类型的B5位点,分别是B5-B和B5-C;而FCC Co纳米颗粒仅表现出一种类型的B5位点即B5-B。同样当配位数为10时,HCP Co纳米颗粒显示三个峰,其中两个峰对应于常规B5-A位,而一个峰对应于B5-B';而FCC Co纳米颗粒表现出两个B5-A位(图7)。
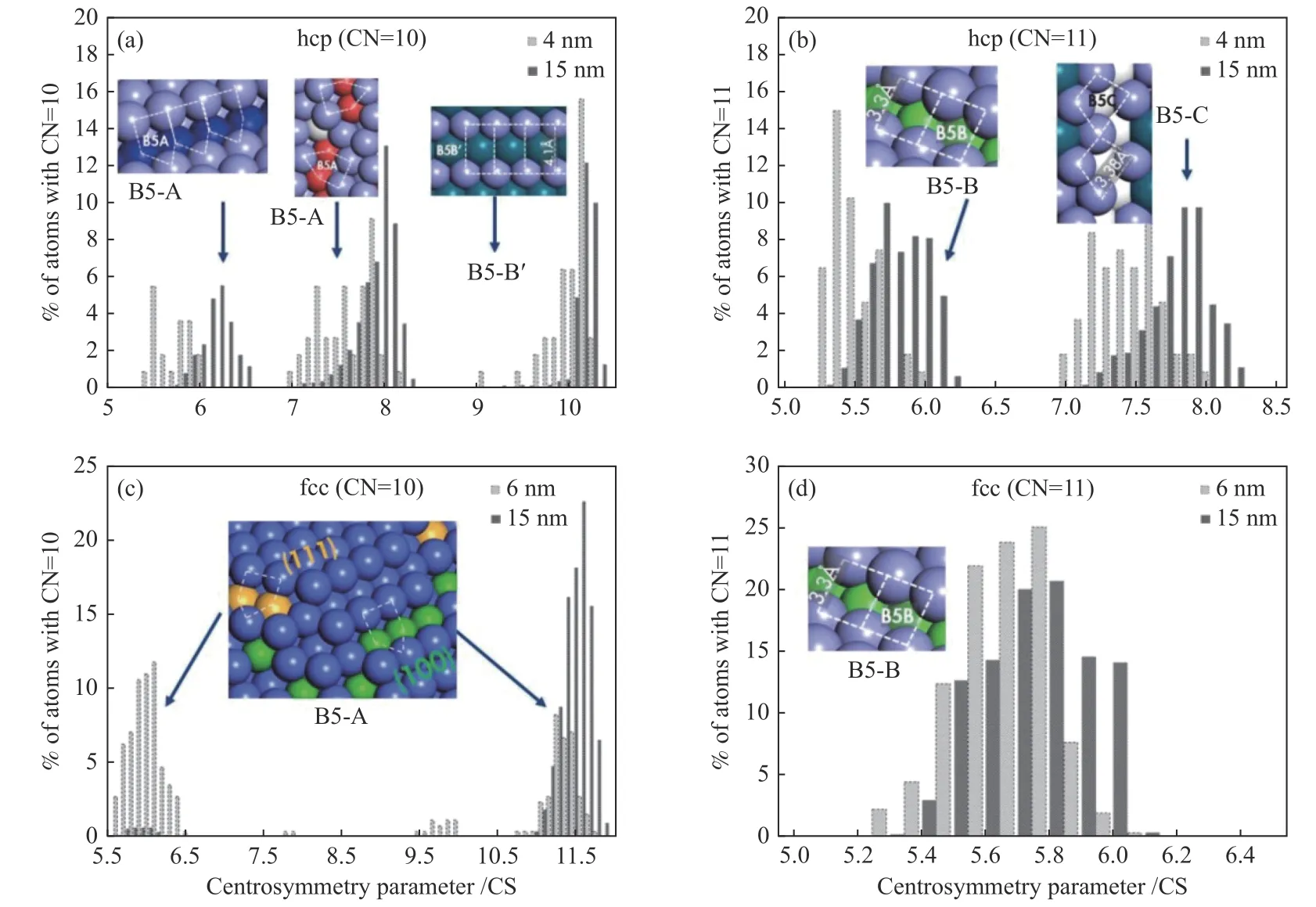
图7 CN=10的 hcp 相和 fcc 相((a)和(c)),CN=11的HCP相和FCC相((b)和(d))[26]Figure 7 Distribution of CS for CN = 10 ((a) & (c)) & CN = 11 ((b) & (d))Top panels are for hcp phase and bottom for fcc phase,Each plot has data corresponding to 2 sizes of nanoparticles as mentioned in the legend,Arrows indicate which distribution corresponds to what type of B5 sites [26]with permission from Elsevier
Wang等[31]研究了Co2C晶面结构特征,从结构性质分析,Co2C(111)和Co2C(001)晶面均有四个吸附位点,分别是顶位、桥位、三重空穴位和四重空穴位;Co2C(101)、Co2C(110) 和Co2C(010)均有三个吸附位点,分别是顶位、桥位和三重空穴位。同时观察到在Co2C(111)和Co2C(001)晶面上的B5活性位由五个钴原子组成,并在CO直接解离上表现出较高的活性。类似地,Liu等[32]在Co、Co2C、Co3C这三种钴相上对FTS反应中决定低碳烯烃选择性的固有位点进行了研究,Co(111)的平坦梯形位包含五个Co原子点作为CO解离的活性位点;Co2C(111)的谷型位点包括四元正方形和三元三角形,作为CHx*物种生成的活性位点,并且这种谷型结构在几何上阻碍了两个相邻CH2之间的耦合;Co3C(101)的山脊型位点包括三个三元三角形,导致CH2*吸附结构的不同,减弱了相互之间耦合的位阻。
Ralston等[9]合成了直径为4.3和9.5 nm的Co纳米粒子,研究发现,在反应过程中存在于表面的单体均被识别为单一碳物种,而主要的CO解离位点则被识别为B5-B位点,了解到Co纳米颗粒活性的差异是由于在较小的纳米颗粒尺寸下这些特定类型位点的损失引起的结构敏感性的结果。另外,van Helden等[33]研究发现,随着纳米颗粒尺寸的增加,B5-B位点的浓度继续增加,CO解离位点的增加导致更高的覆盖度值,从而导致费托反应催化剂有更高的活性。Rankin[34]通过比较Co、Os、Ru的(10-16)表面上小的单原子和双原子物种吸附能的趋势和差异,结果表明,在B5-A和B5-B的台阶边缘位点上,吸附能的差异达到1 eV或者更大,对于几乎所有表面和吸附物,B5-A和B5-B型台阶边缘位点在能量上要优于附近的平台位点。Petersen等[19]对CO直接解离的研究证明,与紧密堆积的Co(0001)和Co(111)表面相比,Co(221)表面B5型位点上的CO直接解离在动力学上更有利。因此,在具有B5型活性位点的催化剂表面,CO倾向于直接解离,从而促进CO活化。
1.4.3 界面位点
催化剂的晶相、晶面、微观活性位结构以及颗粒尺寸一直以来都是影响催化活性的重要因素,界面性质对催化反应性能、产物分布以及催化反应活性也会有影响。Prieto等[35]认为,在Co-SiO2界面处形成的Coδ+位点是导致小粒径钴TOF值降低的原因。在扁平的小钴纳米颗粒中,这些界面部位的相对浓度较高导致d(Co0)<10 nm时观察到的TOF降低。Gnanamani等[36]提出在Co-CeO2界面处部分还原的二氧化铈(Ce3+)和桥接OH参与形成含氧化合物,从而使其选择性增加。
长期以来合成气选择性转化到含氧化物,用的大多都是贵金属,利用非贵金属过渡金属合成各类产物在多相催化中具有重要意义。Liu等[32]认为,Co/Co3C界面与金属Co或Co3C相比,由于电子在界面处的积累促进了电子从界面向吸附的CO*反键轨道的反馈,在CO解离上更具有优势,且该界面对轻质烯烃的形成具有重要意义。Pei等[37]通过理论计算,发现碳化钴对于CO的非解离吸附很高的效率,表现出类似贵金属的特点,而金属钴对于CO的解离吸附和碳链增长具有很高的活性。因此,由金属钴和碳化钴形成的界面以及在Co2C和Co界面形成的“双中心活性位”,可以有效地形成含氧化物,利用双物相催化剂调控催化反应活性,从而来优化合成气转化为含氧化物中非贵金属过渡金属催化剂的设计(图8)。
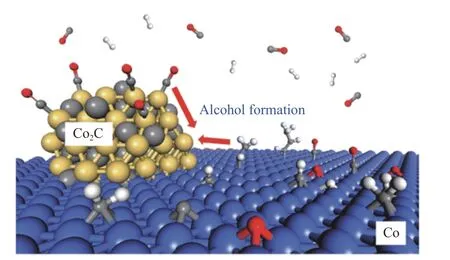
图8 Co-Co2C界面在合成气转化为含氧化物中的应用[37]Figure 8 Interface between the cobalt metal and its carbide phase metal for synthesizing oxygenates in syngas application[37]with permission from ACS Publications
钴基催化剂助剂能改善催化剂的寿命、强度和稳定性,这是因为助剂部分覆盖了催化剂表面活性相,使得化学吸附容量减少,并在金属−助剂界面上产生新的活性位点[38]。Haddad等[39]认为,在以La2O3为助剂的Co/SiO2催化剂中,La2O3形成类岛结构,La3+的存在可能加强了La3+-Co界面附近的烯烃的再吸附,从而提高了链增长概率。Johnson等[40]研究了Mn作为助剂添加到Co/SiO2催化剂中的促进作用,发现随着助剂Mn的增加,其在催化剂表面分布均匀,形成丰富的界面位点,通过STEM-EDS获得的元素谱图的定量分析显示,随着助剂Mn含量的增加,产物的选择性迅速提高。同时研究了Zr作为助剂对钴基费托合成性能的影响,发现Co−Zr界面的存在促进了CO的吸附与解离,有利于费托合成反应活性和C5+选择性的增加,并且抑制了甲烷的生成[41],这与Piao等[42]等的研究结果一致。
1.5 微观活性位的吸附行为
在实际压力条件下的瞬态研究表明,在FTS相关的反应条件下,催化剂表面在很大程度上被一些吸附物覆盖,诸如Had、COad、CHx、OHx和CxHy等。借助各种表征手段和理论计算,了解在费托合成反应条件下形成的表面物质的性质,对最终揭示CH4生成和链增长不同的反应性的表面位点有重要意义。
关于Had在催化剂表面的吸附对催化剂结构的影响,在Lewis等[43,44]的最新研究中,用STM来研究沉积在Cu(111)基底上的两层高的六方晶系的Coad岛上的Had/CO共吸附。该研究在较低的表面温度下进行,证实了Had和CO的分离出现在密堆积的钴表面上。同时研究还表明,被Had部分覆盖的CO吸附在表面上会导致Had吸附物层的压缩,最终导致形成局部高密度(1×1)-1 ML Had结构。
COad对费托合成钴基催化剂的结构和反应性的影响也有大量研究。Lin等[45]研究发现,进料中掺入的CO2会削弱用于CO插入的COads的覆盖范围,从而导致对含氧化合物的选择性降低。许多学者利用表征手段对COads在实验过程中的动态变化进行准确监测,Gunasooriya等[46]对Co(0001)上的COad的结构和电子性质进行了详细的光谱研究,Zijlstra等[18]进行的STM研究表明,COad已在室温下诱导了Co(10-12)表面上Co表面原子的迁移。众所周知,CO吸附在不同的表面,其稳定吸附构型和吸附能也不同。比如CO吸附在平台和缺陷较多的Co表面,特别是CO吸附在顶部位置,会影响COad在各种可能的吸附位置的分布。Paredes-Nunez等[47]进行了从原料气中去除CO的化学瞬态实验,结果发现,存在各种COad(比如线性吸附和桥位吸附),均表现出显著的均一反应性。Zhang等[48,49]发现CO在Co(10-11)、Co(10-10)和Co(11-20)表面分别吸附在4-fold、hcp和4-fold位,对应的吸附能分别为−1.87、−1.77和−1.76 eV。
其他类型的吸附物对钴基催化剂的碳链增长机理和CO解离等方面也有影响。在Wang等[31]使用密度泛函理论计算和微动力学模型研究了在Co2C(101)、(011)、(010)、(110)和(111)面上的CO活化过程。结果表明,CH单体是五个Co2C面中用于CO活化的最丰富的表面CHx物种,并且CO活化和随后的CH形成的机理在很大程度上取决于Co2C面。CH单体的形成速率遵循(111)<(010)<(110)<(101)<(011)的顺序。Floto等[50]对钴纳米颗粒和钴膜进行TPD实验,将两者均先暴露于1 L的H2,然后暴露于1 L的CO气氛当中,观察到钴纳米颗粒的高温脱附峰在800 K,而钴膜的高温脱附峰在660 K,认为是C*和O*与钴纳米颗粒的相互作用更强引起的,也就是说C*和O*将强烈在钴的丰富的台阶-边缘位点结合。
1.6 微观活性位的表征
从结构和电子性质等角度对微观活性位点进行深入分析,了解活性部位的性质和结构-性能关系,均可以帮助设计更高效的催化剂来优化产物的选择性以及更好地揭示活性位点的性质。同时加强在实际催化条件下对催化过程进行原子尺度的研究,利用原位表征技术的应用对深入探究催化反应机理提供参考。
Boeller等[27]提出了一个现场原位扫描隧道显微镜研究,结合实验数据表明,表面台阶就是F-T合成反应中Co(0001)模型催化剂上的活性位点。多种表征手段的结合对催化反应中微观活性位点结构和活性中心的确认提供了可靠的手段。Piao等[42]基于原位/非原位原子分辨STEM成像、EDS元素图谱、电子能量损失谱(EELS)等多种表征技术,揭示了ZrO2助剂含量低时以单分散的形式存在于Co纳米颗粒和和SiC载体上,在Co-ZrO2界面上充当CO解离的真正活性位点。原位EELS分析和密度泛函理论计算表明,Zr原子倾向于结合在钴纳米粒子表面,而不是嵌入到晶格中,同时Zr物种向钴纳米粒子发生电荷转移,从而促进了Zr与Co之间更强的相互作用,从而增强了与H2分子的吸附和CO的解离。2018年Singh等[51]使用原子沉积技术和常规技术相结合合成了Pt-Co双金属催化剂,通过结合反应测试、X射线衍射、电子显微镜和原位红外光谱实验,在密度泛函理论计算的支持下,研究发现了该催化剂表现出对甲醇和低分子量碳氢化合物的选择性的增加,以及对高级醇的选择性略有增加。原位红外光谱测量表明,这些选择性的变化是由于催化剂表面的线式和桥式一氧化碳构型之间的相互作用。首次使用原位DRIFTS实验证明了不同CO结合位点在Pt-Co氢化中的作用。该项研究所采用的方法可用于双金属催化体系对催化机理的深入探究。
费托合成反应是一个低温有利的强放热反应,一直以来希望开发出能够在低温环境下具有高活性特点的钴基催化剂。Wang等[52]提出了一步加氢-还原合成Pt-Co纳米颗粒的方法,该催化剂在433 K下是液相费托合成的优良钴基催化剂。为了探究该双金属催化剂真正的活性位点,在基于原子分辨的球差矫正扫描透射电镜和同步辐射X射线吸收精细结构光谱表征基础上,以CO分子的活化为探针反应,展开了大量的理论计算研究,最终确定Pt-Co双金属催化剂的活性中心为外延生长在Pt纳米粒子表面上的2−3原子层厚的金属Co薄膜。
在多相催化反应中,活性位点在工作条件下结构的动态演变一直是催化领域研究的热点。Weststrate等[53]使用高分辨率发射光谱、动态功函数测量和红外吸收光谱手段,根据表面温度监测CO吸附过程。当 θCO<0.3ML, XPS、FT-IR数据显示,CO仅在顶部吸附并最终形成有序的结构。当 θCO>0.3ML, COad开始在三重空穴位的空心位置填充,此过程伴随着占据顶部位点的减少。Mcnab等[54]利用FT-IR来研究Co/SiO2催化剂上烃类的位置,在Co/SiO2上观察到的碳氢化合物的定量分析表明,更长链物质存在于Co金属本身。大多催化剂的表面结构与反应气氛之间存在动态平衡,催化剂表面会在反应气氛的诱导下发生重构,即催化剂表层原子结构往往是动态的,通常催化剂的活性相和一些反应中间体只是存在于反应条件下,因而准原位技术得到的催化结构以及催化机理很可能不一定准确。近些年来,得益于近常压技术的方法,如近常压X射线光电子能谱[55],使得反应气氛下的原位(in-situ)表征催化剂的结构成为了可能。对微观活性位点的表征,对探究催化反应机理提供了一定的依据,从而能够改善费托反应性能。
2 结论与展望
高比质量活性且稳定的钴基催化剂一直是FT合成反应研究过程中不断追寻的目标,为了最大程度地提高钴基催化剂的F-T反应活性和长链烃选择性,人们不断地尝试从活性位的微观结构以及F-T合成反应过程中催化剂的表面结构变化来探究其构效关系。催化剂不同的晶相和晶面以及费托反应过程中催化剂的表面吸附物等都会对F-T合成反应中催化剂的性能有影响。利用钴的不同形貌或不同晶面,借助各种表征手段和理论计算的方法,微观活性位点和反应性能相关联,对揭示费托反应过程中不同的微观活性位点与吸附物、甲烷生成和碳链增长等的关系都有重要意义,同时可获得更深层次的反应机理方面的信息。
费托合成将来的研究趋势将向催化剂结构的精细化、多功能化方向发展,如采用较好的制备手段控制合成单一晶相结构催化剂和暴露优势晶面等,以及如何在反应过程中使这些具有优异性能的微观结构稳定存在,充分提高每一个钴原子的利用效率,制备更加高效的费托合成催化剂。从而将高效提升合成气直接转化为汽油、柴油或煤油等高品位液体燃料的性能,以使天然气、煤炭和生物质等资源得到更为充分合理的利用,获取最大的经济效益。
猜你喜欢
分子催化(2022年1期)2022-11-02
电气技术(2022年5期)2022-05-23
汽车工程师(2021年12期)2022-01-18
绿色科技(2021年21期)2021-11-26
烟草科技(2021年6期)2021-06-24
第一财经(2019年8期)2019-08-26
航空材料学报(2019年2期)2019-04-15
生物学教学(2018年4期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
延河(2017年7期)2017-07-19
