
饲养方式对藏系绵羊粪便微生物结构与功能的影响
2022-04-25 02:50:46张怀霞张莹莹侯生珍贾建磊
西南农业学报 2022年3期
张怀霞,张莹莹,任 昊 ,谢 雯,陈 倩,侯生珍,贾建磊
(1.青海大学农牧学院,西宁 810016;2.青海大学实验室管理处,西宁 810016)
【研究意义】藏系绵羊作为青海地区传统优良品种之一,草原生态环境的变化和国家禁牧舍饲等相关政策的实施使得放牧补饲或舍饲替代原有的传统自然放牧成为必然趋势[1]。面对牧区草地资源在高寒地带、天然草场具有很长的枯草期等诸多制约条件,只有利用现代养殖技术进行规模化、集约化的饲养管理才能有助于提高藏羊生产效率从而保证牧民养殖的经济效益[2]。【前人研究进展】众所周知,动物胃肠系统的生理动态平衡主要靠微生物来维持,一旦平衡失调,动物的生产性能随之降低甚至发病。研究发现,动物的品种、年龄、日粮以及养殖方式等都在影响着其肠道微生物的结构及其数量。石鹏君等[3]在研究山羊肠道菌群结构组成与物种之间的关系中发现了相同品种的山羊肠道微生物构成相似度较高,不同种类间的山羊之间其肠道菌群构成相似度极低,差异较大;周小娟等[4]在研究不同日龄、品种和饲养方式对肉仔鸡肠道菌群的研究报道中说明肉仔鸡肠道微生物区系受品种和饲养方式等的影响;李贺[5]在探究不同来源脂肪对大鼠肠道微生物的影响研究中发现大鼠盲肠内容物中的菌群发酵和菌群组成受到不同脂肪的显著影响。大量研究表明,动物在不同的饲养模式下其生产性能、微生物区系等都表现出不同程度的差异,例如陈粉粉等[6]在对不同的饲养方式对肉鸡生产性能及肠道和器官指数的影响报道中发现放牧组肉鸡和笼养鸡日增重、血液中的尿酸及总蛋白等均有差异;郑青[7]在不同的饲养模式对猪肠道菌群的影响的研究中发现,林下养殖的猪与圈养猪肠道菌群差异明显且林下散养的猪肠道菌群更具有多样性;王柏辉[8]等以苏尼特羊为对象、以不同的饲养方式为单因素的试验结果发现苏尼特羊在2种饲养条件下其肠道菌群在属水平上表现出极大的差异。【本研究切入点】对于牛、羊等反刍动物,更多的有关其体内微生物的研究都集中在瘤胃,基于反刍动物特殊的消化道结构,其肠道微生物群落也一样值得关注,特别是粪便微生物,往往认为与机体的营养、健康等密切相关[9]。【拟解决的关键问题】以藏系绵羊为研究对象探索不同饲养方式对其粪便微生物菌群结构和功能的影响,旨在为藏羊全舍饲养殖提供理论基础并为藏羊高效养殖提供数据支持。
1 材料与方法
1.1 试验设计
1.1.1 试验动物 试验在青海省共和县切吉乡哇玉香咔牧业责任有限公司(海拔3200 m)进行,选择初始条件相近的1~1.5岁藏羊后备母羊12只,随机分为2组(自然放牧组RF,全舍饲组RS),每组6只。试验从2020年7月8日至9月8日,为期60 d,前2周为适应期,第3周开始为正式期。
1.1.2 饲养管理 试验开始前所有羊只采用传统的放牧+补饲的饲养模式,即藏羊早上8:00放牧,下午5:30归牧,归牧之后补充精饲料和干草。开始后按照试验设计进行羊只的饲养,即放牧组藏羊在当地天然草场自由放牧,早出晚归,归牧后不进行补充饲喂。舍饲组藏羊采用全舍饲的饲养模式,试验期间每天用割草机去草场采割青草带回进行饲喂,自由饮水。
1.1.3 样品采集 饲养试验最后5 d为样品采集期(前2 d为适应期,后3 d为粪便采集期),每只母羊套上粪袋,每天8:00、12:00、16:00和20:00分别随机采集粪便样品,-20 ℃保存,采样期结束后将样品混匀后,每份粪样采集约20 g,放入液氮后带回实验室,-80 ℃保存备用。
1.2 试验方法
1.2.1 样品DNA的提取 按照DP328粪便DNA提取通用试剂盒(天根生化科技(北京)有限公司)说明书提取藏羊粪便细菌总DNA,具体流程如下:①称取180~220 mg的粪样将其放到2 mL的离心管中并将离心管放置在冰上。②在离心管中加入1.4 mL的缓冲液,震荡1 min使样本充分混匀。③70 ℃下孵育5 min。④涡旋15 s之后13 000 r/min离心1 min,然后将1.2 mL上清液转移到另一个新的2 mL的离心管中。⑤加入一个抑制剂吸附片LnhibitEX于离心管中,将其震荡直到吸附片完全并打开重悬。然后在室温下孵育1 min保证吸附片能充分作用。⑥将离心管以13 000 r/min离心3 min。⑦把上一步离心所得的上清液转移到新的1.5 mL离心管后重复步骤6。⑧将离心所得上清液转移200 μL到另一个1.5 mL的离心管后加入15 μL蛋白酶K。⑨加入200 μL缓冲液后涡旋15 s。⑩70 ℃下孵育10 min。加入200 μL无水乙醇并涡旋混匀。把上一步所得溶液加到吸附柱CR2中,12 000 r/min离心30 s后倒掉废液并将吸附柱放进收集管中。将500 μL缓冲液加入到吸附柱CR2中,12 000 r/min离心30 s后倒掉废液,将吸附柱放进收集管中。将700 μL漂洗液加入到吸附柱CR2中,12 000 r/min离心30 s后倒掉废液,将吸附柱放进收集管中。将500 μL漂洗液加入到吸附柱CR2中, 12 000 r/min离心30 s后倒掉废液,将吸附柱放进收集管中。吸附柱CR2放回到收集管后12 000 r/min离心2 min并将废液倒掉,将吸附柱CR2在室温下放置几分钟,以使残余的漂洗液被完全晾干吸附。将吸附柱CR2转到新的离心管中,在吸附膜的中间位置悬空加50 μL洗脱缓冲液,室温下放置2~5 min后,12 000 r/min离心2 min,再将溶液收集到离心管中。
1.2.2 PCR扩增和16S rDNA测序 DNA的纯度及其浓度的检测采用琼脂糖凝胶电泳,离心管内放入一定量的样本DNA,将样本用无菌水进一步稀释到1 ng/μL。将稀释完成的基因组 DNA作为模板,利用16S rDNA V4区通用515F(5’-GTGCCAGCMGCCGCGGTAA-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)PCR扩增目的片段16S rDNA V4区,使用2%浓度的琼脂糖凝胶进行电泳检测;按照PCR产物的浓度开始进行相等含量的混样,充分混匀,然后使用2%的琼脂糖凝胶电泳检测PCR产物,通过胶回收试剂盒来回收相应目的条带产物。接着便构建文库并对其进行定量,定量完成之后选择合格的文库进行下一步的上机测序。
1.2.3 生物信息学分析 测序完成之后,对所得到的原始数据(Raw tags)通过拼接、过滤处理从而得到质量较高的数据(Clean tags)。再将Clean tags去除嵌合体序列之后最终得到样本的有效数据(Effective tags)。
划定97%的相似性为聚类的标准水平,利用Uparse软件对所有样本的Effective tags进行聚类,把有效数据聚类为OTUs,然后用Mothur方法与SILVA132的SSU rRNA数据库对其序列进行物种注释。通过物种注释分析得到每个分类水平上所有样本的群落组成信息,然后对所有样本的数据通过均一化处理,使用R软件进行Alpha多样性分析、主坐标分析、OUTs丰度分析以及功能预测分析,随后得到样本物种丰富度以及菌群多样性等信息,并进一步比较分析不同分组的藏羊其粪便微生物结构和功能的差异。
1.2.4 数据处理 用Excel 2010对数据进行录入整理,采用SAS 9.0对数据进行统计分析,以α=0.05进行显著性检验,数据均采用平均值±标准差表示。
2 结果与分析
2.1 测序质控结果
利用Illumina Nova 测序平台进行测序,建立PCR-free文库,随后开始双末端(Paired-end)测序。对Reads拼接之后,12份藏羊粪便样品平均每样品测得91 291条tags,经过质控平均得到 86 365条有效数据,质控有效数据量达60 408条,质控有效率达66.03%。各样品测序质控数据统计见表1。
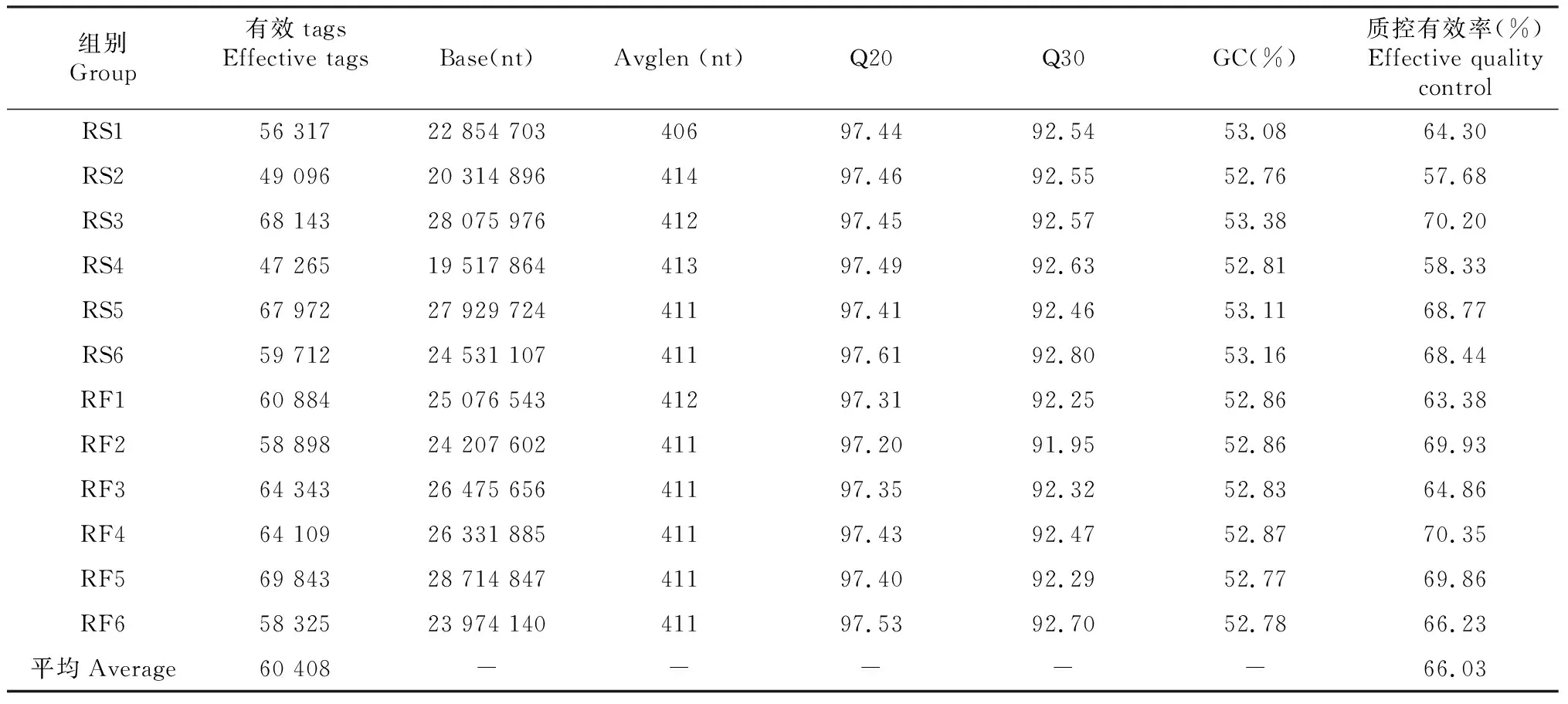
表1 数据预处理统计及质控
2.2 组内同质性分析
由图1可知,在放牧饲养条件下藏羊粪便样品中检测到的OTU数目最多为1787个,舍饲饲养条件下则检测到1630个OTU。舍饲组6个样本共有的OTUS数目为812,放牧组为489,说明放牧组的组内同质性较舍饲组好。
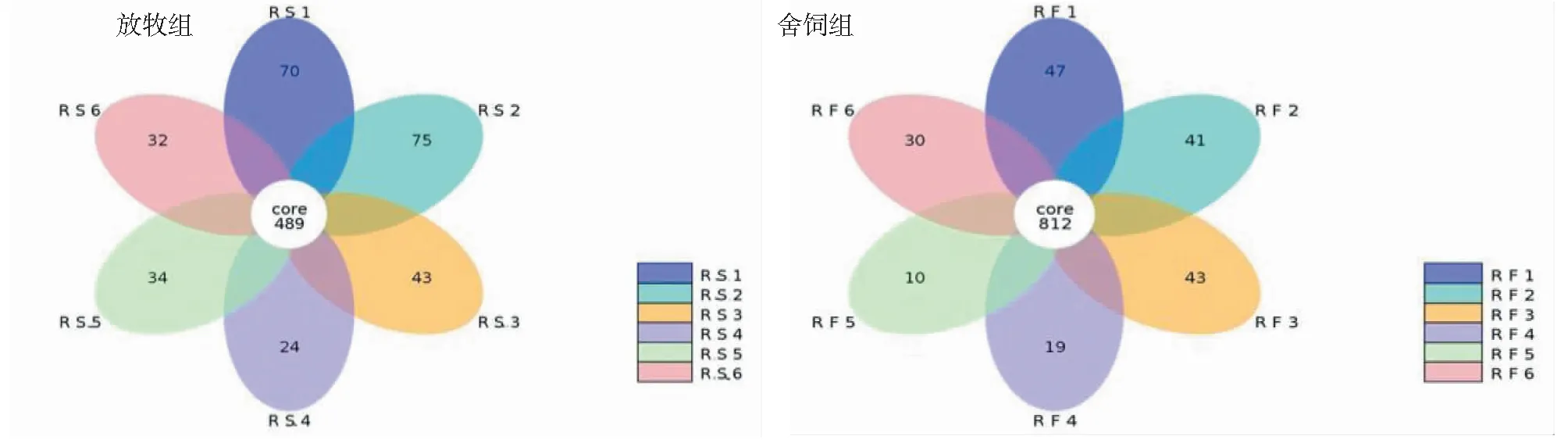
一个完整的花即为一组样本,而不同颜色的花瓣代表组内不同样本。core数字反映了所有样本共有OTUs数目,花瓣上的数字为该样本(组)独有的OTU数目A complete flower is a group of samples, and petals of different colors represent different samples within the group. The core number reflects the number of OTUs shared by all samples, and the number on the petals is the number of OTUs unique to this sample (group)图1 各样本同质性分析Fig.1 Analysis of homogeneity of each sample
2.3 Alpha多样性分析
将不同样本在97%的相似度水平下获得的Alpha Diversity分析指数进行统计,结果如表2所示。其中,Observed-species指数反映的是群落中物种的数量,结果表明放牧组样品中群落的物种数量显著高于舍饲组(P<0.05);Shannon指数则反映菌群的多样性,数据表明2个组的样品菌群多样性差异显著(P<0.05),说明放牧组藏羊的粪便样品其菌群较舍饲组具有更高的多样性。
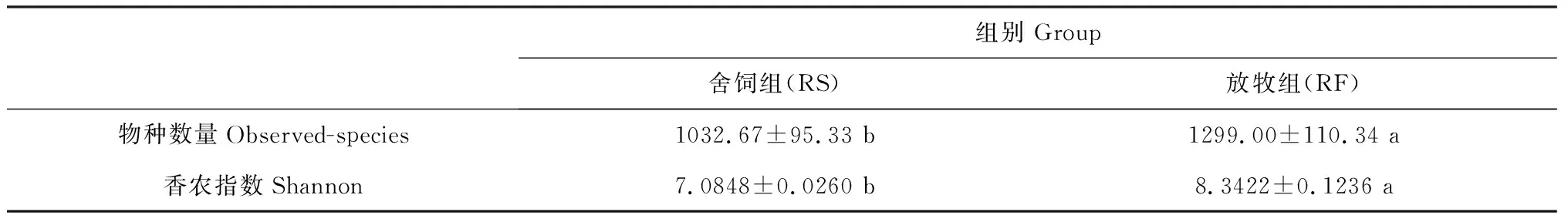
表2 Alpha多样性统计
2.4 物种注释及OTUs丰度分析
通过物种注释并对不同分类层级统计发现共有1928个OTUs,其中,能够注释到数据库的OTUs数目为1927(99.95%),注释到界水平的比例为99.95%,门水平的比例为98.08%,纲水平的比例为96.21%,目水平的比例为92.74%,科水平的比例为80.86%,属水平的比例为27.96%,种水平的比例为8.04%。
2.4.1 菌群在门分类水平上的比较 如图2所示,基于门水平而言,每一个样品中占据主导地位的主要包括厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)和变形菌门(Protebacteria)。其中厚壁菌门和拟杆菌门这两大门类序列占总序列的75%以上,是藏羊粪便微生物菌群中的绝对优势菌。结合表3来看,舍饲组样本中的放线菌门(Actinobacteria)和螺旋体门(Spirochaetes)的相对含量显著高于放牧组(P<0.05),其他菌门在2组中均差异不显著(P>0.05)。
2.4.2 菌群在科分类水平上的比较 由图3可知,在科水平上以瘤胃菌科(Ruminococcaceae)、毛螺菌科(Lachnospiraceae)和Unidentified-clostridiales的丰度最高,即为优势物种。
进一步分析科水平上的细菌丰度(表4),其中放牧组在科水平上克里斯滕森菌科和拟杆菌科的含量显著高于舍饲组(P<0.05),其余菌科在2组中均有差异但不显著(P>0.05)。从表中可以看到,2组中除了瘤胃球菌科、毛螺菌科及未被分类梭菌科之外,理研菌科和克里斯滕森菌科也具有较高的含量。
2.5 主坐标分析
PCoA分析说明,如果样本都聚集在一起,说明它们的物种结构组成有很高的相似性,而如果群落的差异性很大,样本就会分开且距离较远。由图4可知,不同饲养模式下饲养的藏羊样本菌群聚类在坐标不同的位置,组间差异明显而组内没有明显的差异,舍饲组样本中的物种组成结构相似度较高,表明不同饲养模式对藏羊粪便微生物群落有影响。
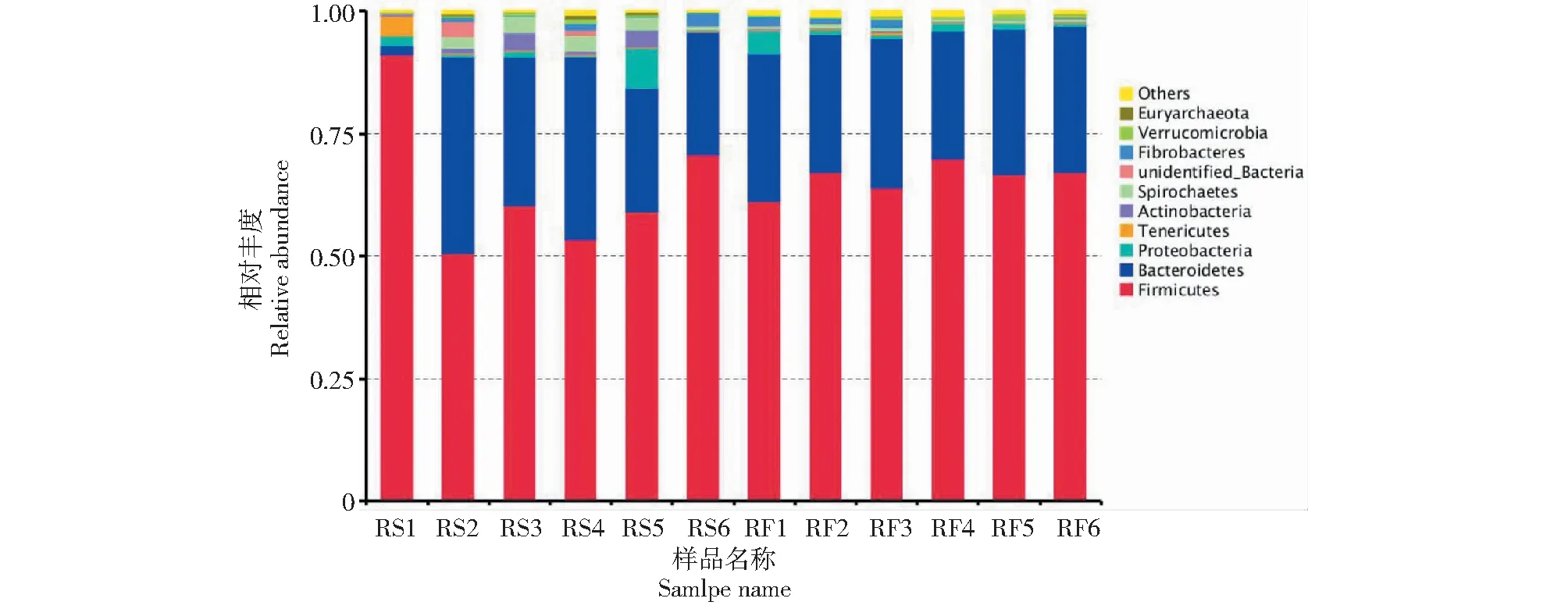
图2 门水平上物种分析Fig.2 Species analysis at the phylum level

表3 门水平上的细菌相对丰度
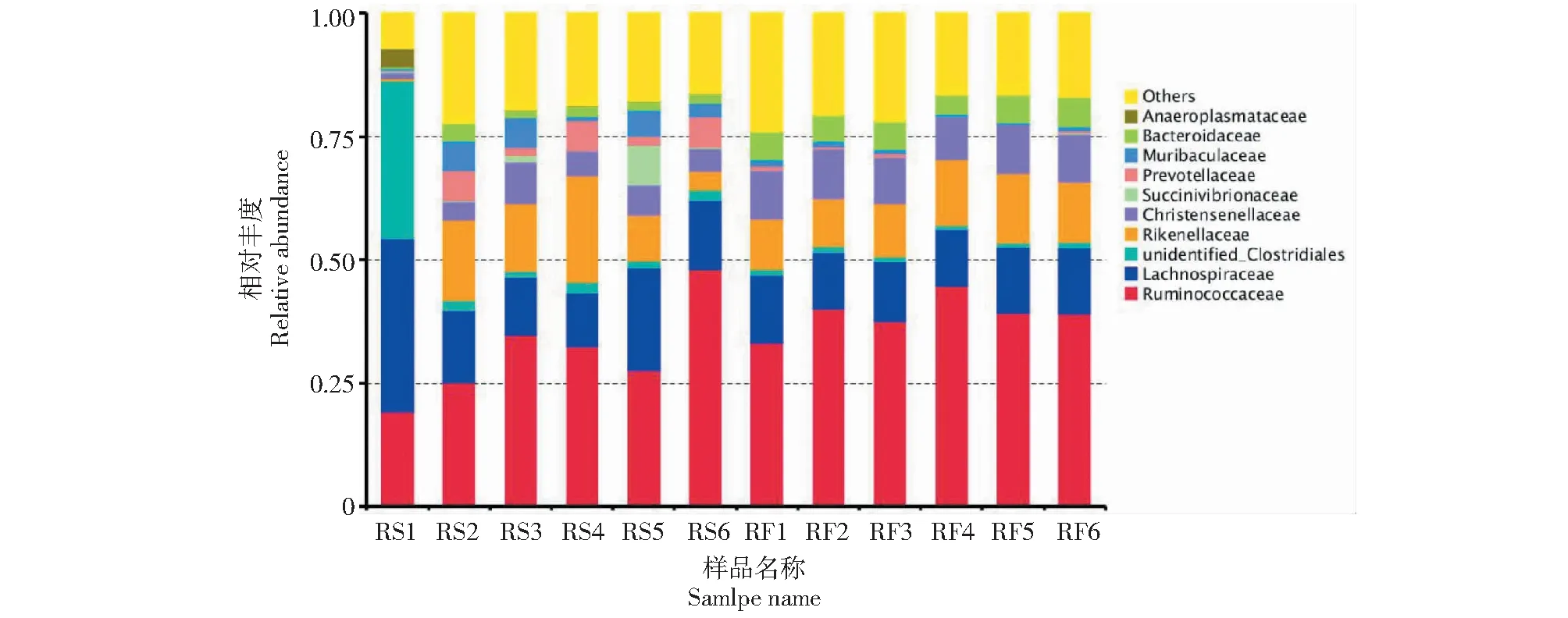
图3 科水平上物种分析Fig.3 Species analysis at the family level
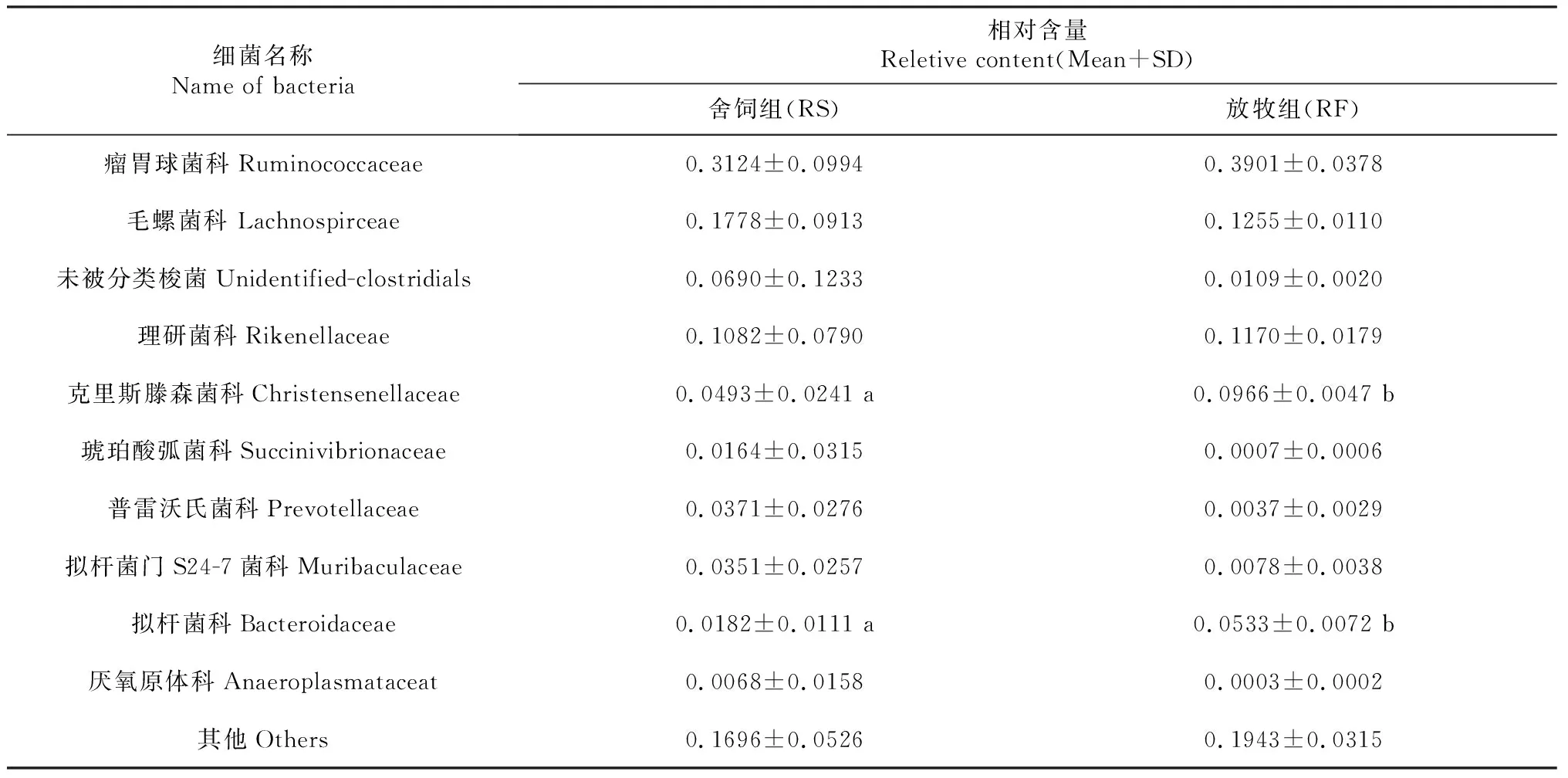
表4 科水平上的细菌相对丰度
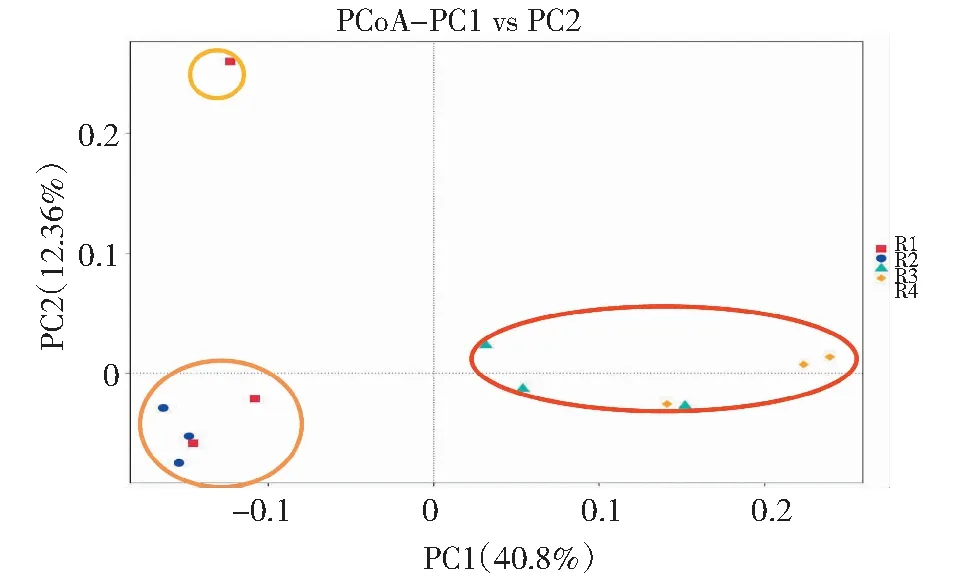
R1:舍饲组1; R2:舍饲组2; R3:放牧组1; R4:放牧组2R1:House feeding group 1; R2:House feeding group 2; R3:Grazing group 1; R4:Grazing group 2图4 Unifrac加权主坐标分析Fig.4 Unifrac weighted main coordinate analysis
2.6 功能预测分析
选择每个样本在每个注释层级上最大丰度排名前10的功能信息,作出功能相对丰度柱状堆积图进而查看各样本在不同注释层级上相对丰度较高的功能及其比例。如图5所示,2个组的藏羊粪便微生物功能所占比例差异不明显,在2种饲养模式下藏羊粪便微生物的功能主要集中在新陈代谢(Metabolism)、基因信息和环境信息的加工(Genetic-information-processin)。
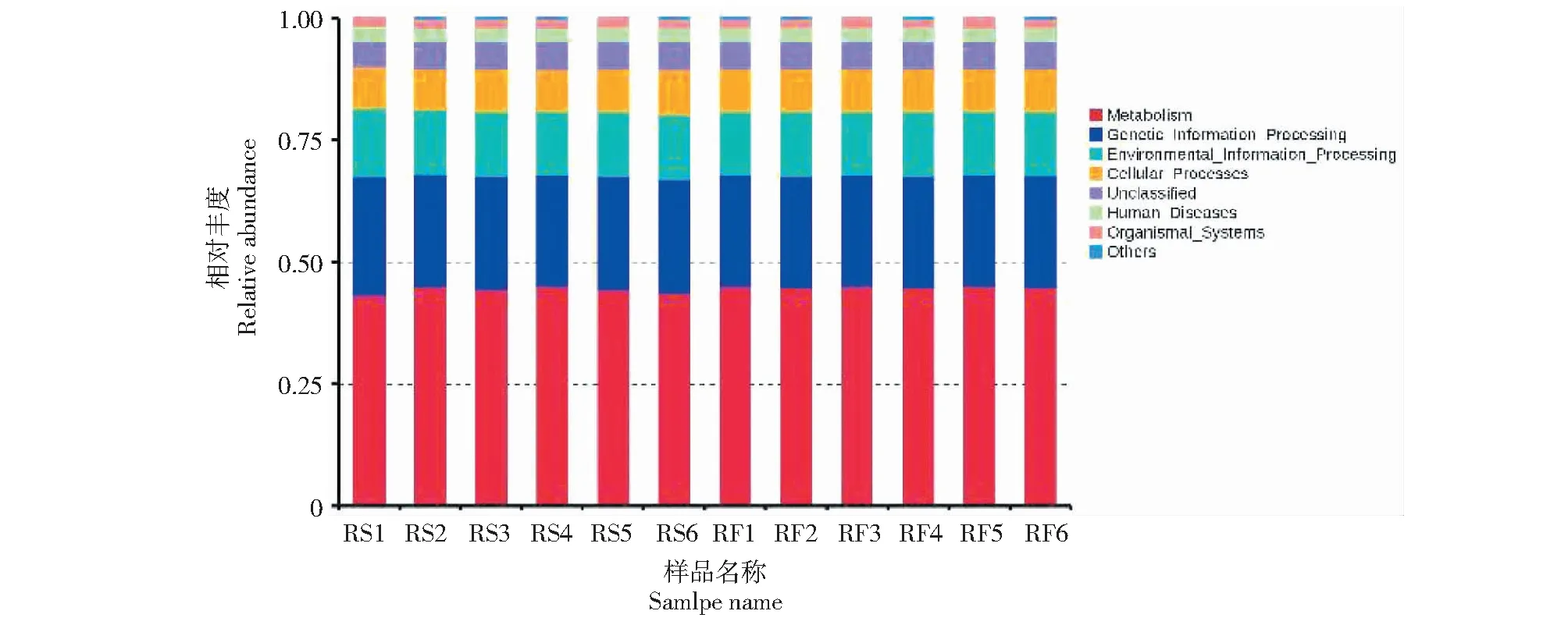
图5 KEGG功能注释相对丰度Fig.5 KEGG feature noterelative relative abundance
3 讨 论
3.1 不同饲养模式对藏羊粪便微生物群落结构组成的影响
从测序结果发现藏羊粪便中微生物的种类丰富,其粪便微生物菌群包含21个门,30个纲,46个目,82个科和161个属的细菌群落。本试验在放牧饲养条件下的藏羊粪便样品中检测到的OTUs数目为1787个,舍饲条件下则检测到1630个OTUs,结合Alpha多样性分析结果表明放牧组藏羊比舍饲组藏羊粪便菌群更具有多样性。
基于门分类水平而言,藏羊粪便样品中厚壁菌门、拟杆菌门和变形菌门的丰度最高,为主要的优势菌,这和多数研究结果[10]类似。在门水平上舍饲组样本中的放线菌门和螺旋体门的含量显著高于放牧组(P<0.05),放线菌门作为大多数哺乳动物肠道微生物的主要优势菌[11],能够产生大量的抗生素消灭致病菌群从而保护宿主,也可以产生蛋白酶有利于宿主对蛋白的消化吸收[12],而螺旋体门作为致病菌在舍饲组样本中含量较高,证明可能是较高含量的螺旋体导致舍饲组一号样本与组内其他样本产生了明显的差异。2组藏羊样本粪便微生物菌群科水平上克里斯滕森菌科和拟杆菌科丰度均有显著差异(P<0.05),两者均在放牧组中具有较高的丰度,说明放牧组的藏羊对能量的利用率较高。试验中的以上发现都说明不同的饲养模式对藏羊粪便微生物群落结构组成有影响,许多研究也都证明了不同的饲养模式对动物粪便微生物的影响:杨伟平[13]关于放牧型藏猪与普通瘦肉型猪肠道的细菌群落组成及多样性的研究表明,藏猪肠道菌群多样性显著高于普通的瘦肉型猪,这是因为藏猪长期放牧采食牧草和幼虫等;孔令玲[14]探究雪山鸡在不同饲养模式下肠道微生物的差异时发现雪山鸡在后期的笼养与全程平养2种饲养方式下物种丰度和菌群多样性有明显的差异。所以在此试验中藏羊生活习性的改变可能造成其粪便微生物群落结构产生差异包括光照、接触的土壤环境以及藏羊的行为活动方式等。
3.2 不同饲养模式对藏羊粪便微生物功能的影响
动物肠道中栖居的大量微生物和胃肠组织构成一极其复杂、对机体健康起着重要作用的菌群系统。由于基因组学的不断发展,动物粪便微生物的功能也被逐渐地发现,细菌和宿主通过物质和能量以及基因的互动来调节重要的化学转化[15]。本试验结果表明,藏羊粪便微生物的主要功能包括新陈代谢、基因信息和环境信息的加工等,这可能主要取决于藏羊肠道内丰度较高的细菌。宿主碳水化合物、蛋白质及其他物质的降解离不开肠道内的拟杆菌,而膳食纤维的降解利用也要依赖于肠道内的厚壁菌[16]。试验发现科水平下瘤胃球菌科和毛螺菌科含量最高,瘤胃球菌科可以产生短链脂肪酸,其与反刍动物纤维素等多糖的消化相关;毛螺菌科可分解纤维,包括试验检测出的克里斯滕森菌科和琥珀酸弧菌科等都在宿主的新陈代谢中发挥自己的功能作用,它们可以通过消化纤维素及半纤维素,分解从而产生多糖[17]。在科水平下,克里斯滕森菌科和理研菌科都具有较高的丰度,其中克里斯滕森菌科一直都与基因联系在一起,有研究提出克里斯滕森菌科是遗传度最高的肠道菌;理研菌科与宿主的健康有着紧密的关系,主要表现在它在动物肠道内的含量会影响肠道内短链脂肪酸即丁酸和戊酸的含量,从而对动物健康产生影响[18]。
通过KEGG功能注释发现,不同饲养模式下藏羊粪便微生物功能没有显著差异,具有较好的一致性,可能说明不同的饲养模式改变了藏羊粪便菌群的结构及其多样性,但对微生物功能的影响并不明显,可能进一步表明藏系绵羊适合舍饲饲养。
4 结 论
通过对比不同饲养模式下藏羊粪便微生物菌群结构与功能,发现不同饲养模式下的藏羊粪便微生物菌群差异显著,藏羊在自然放牧饲养方式下粪便微生物菌群多样性高于全舍饲饲养方式;藏羊粪便微生物功能主要集中在新陈代谢、基因信息处理和环境信息处理中,2组藏羊其粪便微生物功能没有显著性差异;综上所述,不同的饲养模式虽然对藏羊粪便微生物结构影响差异显著,但对微生物功能的影响不明显,表明藏系绵羊能够适应全舍饲饲养模式。
猜你喜欢
中老年保健(2022年2期)2022-08-24 03:20:50
疯狂英语·新悦读(2021年10期)2021-11-23 03:04:01
科学(2020年4期)2020-11-26 08:27:06
科技视界(2020年26期)2020-09-24 03:25:06
科技视界(2020年17期)2020-07-30 14:03:27
家庭医学(下半月)(2019年9期)2019-10-12 08:03:52
化学教学(2018年1期)2018-02-28 21:26:29
发明与创新(2016年33期)2016-04-16 16:32:25
动物营养学报(2015年10期)2015-12-01 02:26:20
现代检验医学杂志(2015年4期)2015-02-06 02:02:11
