
基于近邻样本联合学习模型的疟疾识别算法
2022-04-22 08:55哈艳孟翔杰田俊峰
河北大学学报(自然科学版) 2022年2期
哈艳,孟翔杰,田俊峰
(1.河北大学 管理学院,河北 保定 071002;2.河北省高可信信息系统重点实验室,河北 保定 071002; 3.河北大学 数学与信息科学学院,河北 保定 071002;4.河北大学 网络空间安全与计算机学院,河北 保定 071002)
疟疾是一种由疟原虫引起的传染性疾病,有至少7天的潜伏期[1-2].这种疾病每年夺去数百万人的生命,很难被发现和治疗,特别是在缺乏检测设备和医疗专家的偏远地区及发展中国家,因为它的症状与普通感冒相似[1],如果患者没有及时接受治疗,会有一定的生命危险[3].
疟疾在疟原虫感染各个时期的表现不同,因此,对疟疾的多阶段识别在临床治疗中具有重要的实用价值.目前已经出现许多鉴定疟原虫的技术,其中经过染色的血液涂片的显微镜检查是目前最常用的检测方法[4-6].虽然还有其他一些在疟疾检测中表现良好的方法,比如荧光定量聚合酶链式反应技术[7],但显微镜因其成本低、操作简单的优势而成为最受欢迎的检测方法.在没有计算机辅助的情况下,其检测性能基本上取决于检查员的专业水平,这需要耗费大量昂贵的人力资源.因此,通过计算机辅助系统提高显微镜下疟原虫识别的准确性是一个热门的研究领域.
最近,疟原虫自动识别方面已经出现了很多有效的方法,其中大多数是通过CNN来实现的,它从图像相邻像素中挖掘最重要的信息用以判断疟原虫感染的阶段.然而,这些深度学习系统仅研究感染细胞与健康细胞的二分类识别,不能胜任疟疾的多阶段识别任务.
针对上述问题,本文提出了一种新技术——近邻样本联合学习模型(NSJL),它分为3个模块:首先是CNN特征学习,用于提取样本的主要特征;然后,使用K-NN建图算法来挖掘相似样本之间的关系;最后将CNN特征和样本间关系通过GCN进行进一步优化,实现对疟疾寄生虫的多阶段识别.
在疟疾多阶段识别实验中,笔者提出的NSJL模型达到了92.50%的正确率、92.84%的精确率、92.50%的召回率和92.52% F1分数.与对比方法相比,NSJL在疟疾多阶段识别问题中提高了7.0%正确率、5.79%精确率、7.0%召回率和6.83%的F1分数.通过大量的实验,验证了NSJL模型在多阶段寄生虫识别任务中的能力和可扩展性.
1 相关工作
1.1 特征工程
已有的一些研究工作根据图像的特征开展疟疾识别任务,一般是将被检测的细胞图像转换为手工设计的特征向量(包括形态学、颜色和纹理等特征),这样可以更容易让计算机进行后续分类任务[8-10].Lopez等[8]提出利用局部图像描述技术建立可以识别6种类型白细胞的辅助系统,并使用关键点检测器和其他一些常规采样技术组成图像特征提取技术库.此外,还有一些实用的技巧经常被使用,如Markiewicz等[9]和Ross等[10]使用了一些基于手工提取的图像特征,包括但不限于形状、颜色或纹理直方图的分析.虽然研究结果表明使用上述方法是合理的,但这些方法仍然没有结合相邻的图像像素学习更全面的信息.此外,它们只在小数据集上取得了很好的结果,但在大数据集上表现不佳.
1.2 机器学习方法
基于机器学习的计算机自动疟疾识别方法近年来发展迅速,目前已经有许多研究应用机器学习方法来识别疟疾寄生虫[11-13],它们主要包括一些基本的分类算法,比如支持向量机(support vector machine,SVM)[11].其中,Tek等[12]对其数据集进行归一化和颜色校正后,提出了一种改进的K-NN方法进行二分类.Parkt等[13]使用未染色图像的定量分析来检测由恶性疟原虫感染的红细胞.
除了上述传统机器学习方法,能够从数据和标签中自动提取感兴趣的特征的深度学习技术也是显微图像识别的常用方法之一[14-15],与机器学习方法相比它的效果更加优秀.近年来,已经出现许多使用深度学习来进行血液涂片的自动寄生虫识别的研究.Delahunt等[14]在对疟疾样本进行定位后,提出了一种将线性SVM和多个卷积层相结合的疟疾识别方法.它使用CNN进行特征提取,并在患者水平达到了92%的特异性.另外, Mehanian等[15]首先利用高斯核支持向量机对WBCs进行识别,然后使用CNN模型作为特征提取器和分类器,该方法在块水平上的灵敏度为91.6%,精度为89.7%,特异性为94.1%.然而,上述方法仅用于解决二元寄生虫识别问题,不适用于多阶段疟原虫识别.
在本研究中,提出了一种新的用于多阶段寄生虫识别的算法,即近邻样本联合学习模型,它主要有3个关键步骤:首先,自动提取特征表示(CNN特征学习);其次,建立基于相似度的邻域关系(邻域相关性挖掘);最后,通过图卷积网络对CNN特征进行优化(图特征嵌入).
2 方法
2.1 NSGL模型的框架
提出了一种针对多阶段疟疾寄生虫识别问题的近邻样本联合学习(NSJL)模型,该网络由CNN特征学习、邻域相关性挖掘和图特征嵌入3个模块组成.如图1所示的框架方法,首先,将原始图像I={i1,i2,…,ij,…,in}输入到卷积神经网络模块中,得到CNN特征X={x1,x2,…,xj,…,xn}.然后使用K-NN算法,通过CNN特征之间相似性计算邻接矩阵.之后,将邻接矩阵和CNN特征传入图卷积网络进行运算,得到原始图像的最终特征Z={z1,z2,…,zj,…,zn}.最后,训练交叉熵分类器,通过输入GCN特征来优化网络参数.
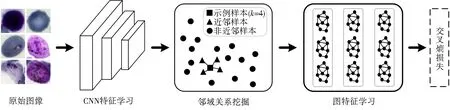
图1 NSJL方法框架Fig.1 Framework of our NSJL
2.2 CNN特征学习
特征学习模块基于ResNet[16]网络,通过添加残差块加深网络结构,是最近最流行的CNN特征提取器.解决了深层网络存在的问题:随着网络深度的增加,网络的效果受梯度爆炸(或消失)以及复杂优化问题的影响不一定会提高.
ResNet由许多残差单元组成,每个单元包括恒等映射和残差连接.具体来说,给定原始图像I={i1,i2,…,ij,…,in},其中ij代表I中的第j张图像,n代表图像总数.残差单元的计算公式可以表示为
(1)
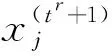
,
(2)
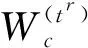
类比其他相关研究,本文选择50层的ResNet网络(ResNet50)作为CNN特征提取器,它主要包括1个单个卷积层,16个残差单元和若干全连接层.通过这个网络将原始数据转化为需要的CNN特征X={x1,…,xj,…,xn}.
2.3 邻域相关性挖掘
建图方法主要通过CNN特征来挖掘样本之间的关系,一个优秀的建图方法有助于算法的后续优化.本文使用K-NN建图算法作为图构建算法.K-NN算法是目前最常见的分类算法之一,它可以通过邻接矩阵量化样本之间的关系,其核心思想是将k个在特征空间中与目标样本最近的图像与目标样本连接起来.
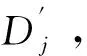
(3)
K-NN建图算法需要计算特征相似度,在本文模型中,特征相似度是用欧氏距离来度量的.
2.4 图特征嵌入
在获得CNN特征集合X={x1,x2,…,xn}及其邻接矩阵A后,使用GCN[17]提取比CNN更抽象的图嵌入特征.GCN的结构分为2个模块,包括若干用于特征提取的图卷积层和一个用于分类的感知器层.图卷积层利用邻接矩阵和CNN特征挖掘样本之间更深入的表示形式,如下式:
(4)
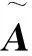
另外,GCN的感知器层定义为
(5)
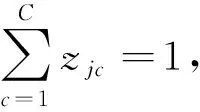
为了优化NSJL模型的可训练参数W={Wc,Wg},将交叉熵损失添加到GCN特征上.
,
(6)
其中yj代表第j个样本的真实标签.NSJL算法的整体流程展示在算法1.
算法1 NSJL
1)输入:原始图像I,批量大小nb,K-NN算法的参数k.
2)初始化:随机初始化CNN和GCN网络的参数集合W,使用全零矩阵初始化邻接矩阵A
FORj=1 TOnbDO
从I中选择1张图像ij.
使用公式1计算CNN特征xj.
END FOR
FORj=1 TOnbDO
使用公式(3)更新邻接矩阵A.
使用公式(5)获得GCN分类结果Zb.
使用反向传播算法利用公式(6) 优化网络参数W.
3 实验
3.1 数据集
为了实现疟疾多阶段识别的任务,使用了从吉姆萨染色液试剂染色的血液涂片上获取的细胞图像作为数据集.它总共包含1 364张图像,在Broad bioimage benchmark collection(BBBC)(https://data.broadinstitute.org/bbbc/BBBC041/)网站上公开[18].3位世界级的专家对这些图像进行了手工注释,数据总共被分为6类,包括2类健康细胞(红细胞和白细胞)和4类感染细胞(配子体细胞、环状体细胞、滋养体细胞和分裂体细胞).数据集提供了细胞的边框坐标和相应的阶段标签,因此可以从中分割出7 256张细胞图像.由于一些类(如白细胞)的样本太少,本文新拍摄了200张图像,并从整个数据集中为每个类随机选择100张图像作为测试集(总共600张图像).总共使用7 456张细胞图像,包括6 856张作为训练集和600张作为测试集.
3.2 实验设置
网络参数设置:NSJL模型包括CNN/GCN网络,并使用Adam优化器通过交叉熵损失对模型进行优化.实验的批量大小设置为70,学习率为2×10-3,每50 次迭代减少为原来的1/10倍.在邻域相关性挖掘模块中设置k=6.NSJL模型的训练采用PyTorch框架在GTX2080 GPU上运行,最大迭代次数为60.
评估标准:通过NSJL模型的平均准确率、精确率、召回率和F1分数来评估在多阶段疟疾寄生虫识别任务中的表现.最后给出了原始的二分类评价方法的计算公式.
(7)
(8)
(9)
(10)
其中,TP、FP、TN、FN分别表示预测正确的正样本数量、预测错误的正样本数量、预测正确的负样本数量、预测错误的负样本数量.
为了进一步验证NSJL模型的性能,还使用了接收者工作特征(receiver operating characteristic,ROC)曲线和t分布随机近邻嵌入(t-stochastic neighbor embedding,t-SNE)图来分析结果.
3.3 结果
为了验证NSJL模型在多阶段疟疾识别问题上的优越性,本文将NSJL模型与其他经典的深度学习分类算法进行了比较,包括VggNet[19]、GoogleNet[20]和ResNet50[16].
表1总结了所有实验的结果,本文的算法的准确率、精确度、召回率和F1分数的值分别为92.50%、92.84%、92.50%和92.52%.与其他分类算法相比,NSJL模型在准确率、精确率、召回率和F1分数上分别超过了它们最好效果7.00%、5.79%、7.00%和6.83%.证明样本间相关关系的算法可以显著提高多阶段疟原虫识别任务的准确性.
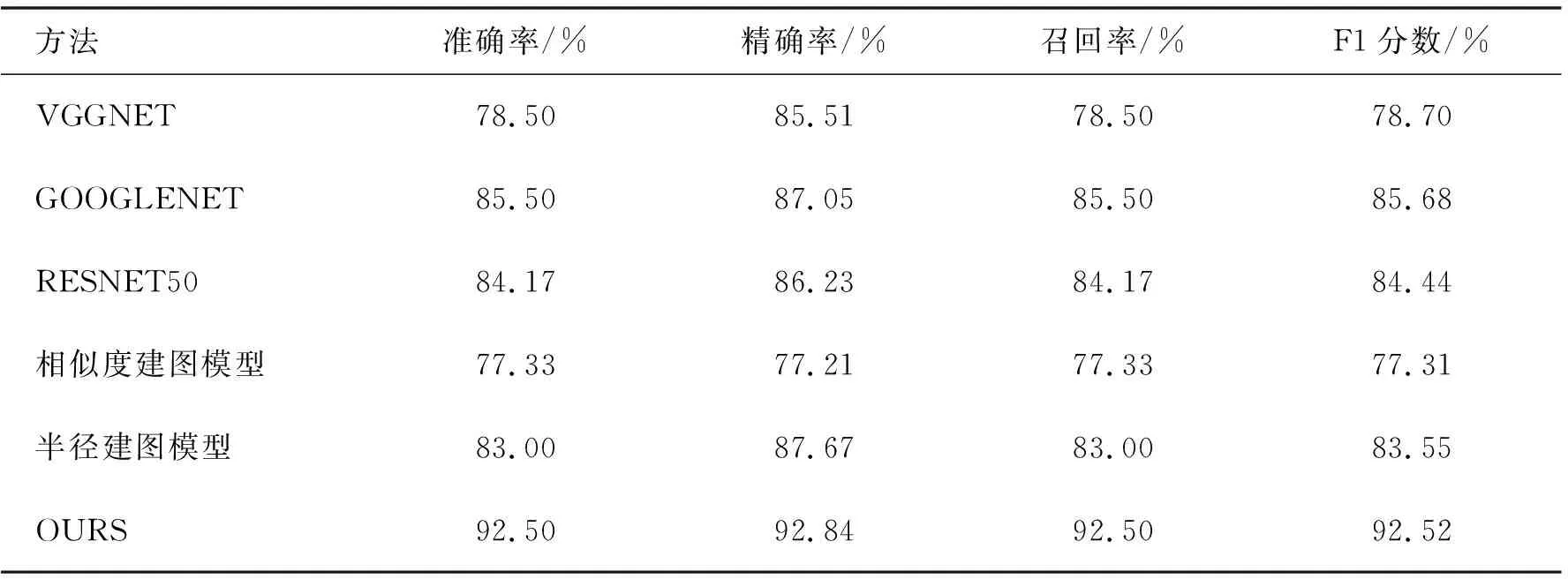
表1 不同方法在多阶段数据集上的结果
此外,ROC曲线和AUC值反映了设计的分类器的鲁棒性,因此使用它们来证明本文的NSJL模型的分类性能.由图2可以看出,NSJL模型在整体分类中ROC曲线表现优异,但在裂殖体阶段的结果较其他阶段较弱.此外,NSJL总体的AUC值达到了0.98,配子细胞、白细胞、红细胞、环状体细胞、裂殖体细胞和滋养体细胞的AUC值分别达到了0.97、0.99、1.00、1.00、0.98和0.96.从ROC曲线和AUC值可以看出,本文的模型在多阶段疟疾识别问题上有出色的性能.
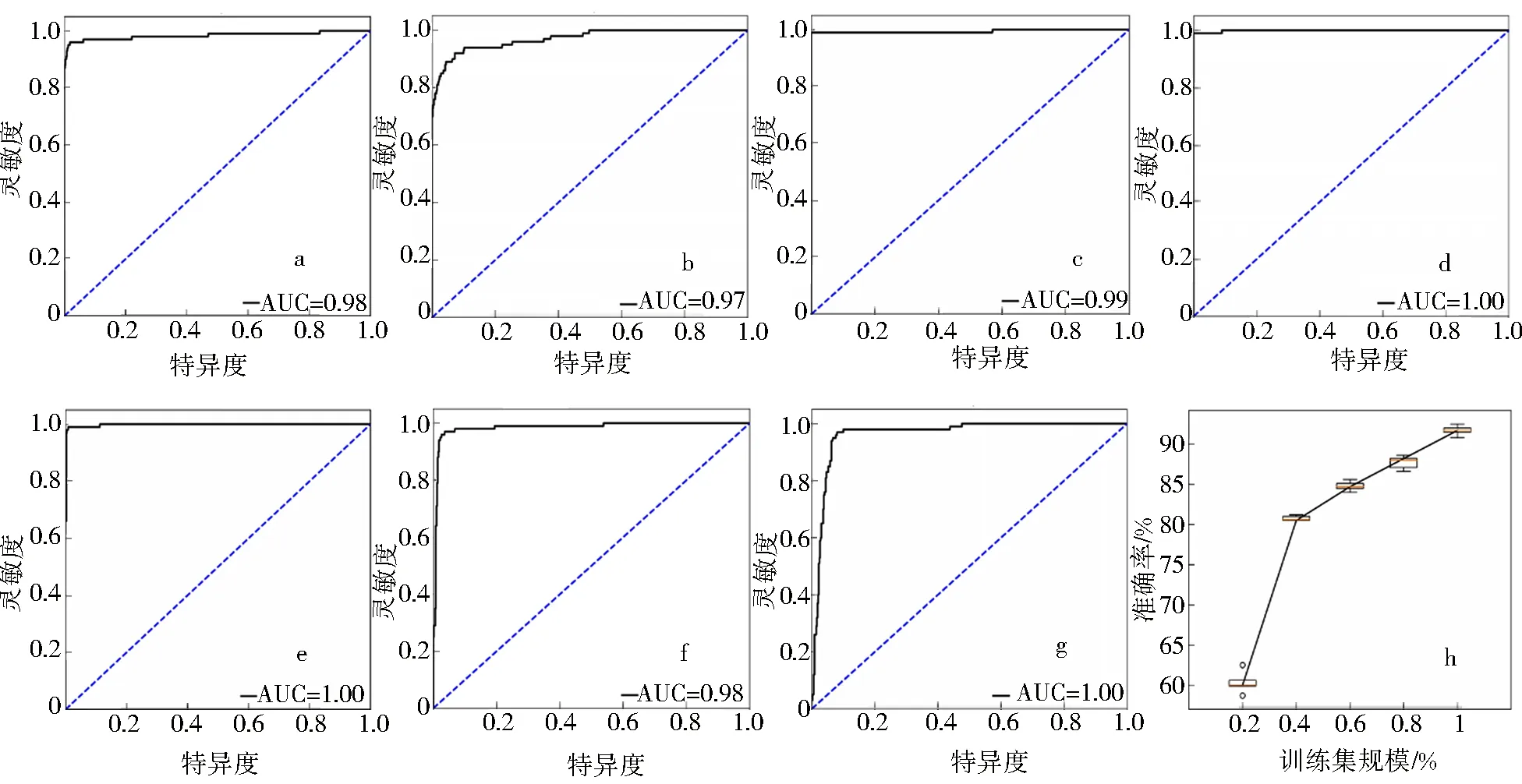
a.总体;b.配子体;c.白细胞;d.红细胞;e.环状体;f.裂殖体;g.滋养体;h.不同训练集规模下的模型效果.图2 NSJL模型的ROC曲线和训练集规模评价Fig.2 ROC curves and training size evaluation of NSJL model
此外,对网络在感知器层之前的输出特征绘制了t-SNE图.该可视化技术可以说明本文的NSJL模型对高维特征的判别能力.因此,绘制了表1中的每种方法的2D t-SNE图来可视化学习到的特征,图3f中的可视化表明,NSJL模型清晰地将每个阶段的特征表示分离开来,来自同一阶段的样本聚在一起.与VggNet、GoogleNet和ResNet50的图3a-c相比,NSJL有着更好的分离效果.这是因为考虑了样本之间的关系和结构,通过这种结构可以更好地优化网络参数.

a.VggNet;b.GoogleNet;c.ResNet50;d.Ours+相似度建图;e.NSJL+ε半径建图;f.NSJL.图3 每种方法的t-SNE可视化Fig.3 The t-SNE visualization of each method
3.4 讨论
本文提出的近邻样本联合学习模型(NSJL)是由CNN特征学习、邻域相关性挖掘和图特征嵌入模块组成的,本部分将对这些模块进行讨论.另外,也在不同的参数和训练数据量下对NSJL的性能进行了评估.
3.4.1 CNN特征学习模块的讨论
为了评估CNN特征学习模块对整体模型的贡献,将ResNet50替换为ResNet18和ResNet34,在不改变其他模块和参数的情况下验证性能.如图4a所示,ResNet50的效果更好,分别提高了27.34%(ResNet18)和36.67%(ResNet34).原因是更深层的网络可以从原始图像中提取更抽象的特征,为图卷积网络提供更充分的个体特征表示.
3.4.2 邻域相关性挖掘模块的讨论
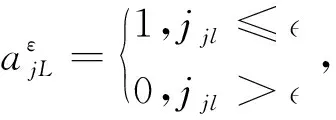
3.4.3 图特征嵌入模块的讨论
NSJL的另一个主要模块是图特征嵌入,这一部分评估了图卷积网络对NSJL的影响.对不同数量的图卷积层开展实验进行对比,获得的结果在图4b中,很容易看到GCN网络深度的变化几乎没有影响本文模型效果,但图卷积层的最佳数量层是2,可达到92.50%的准确率.
3.4.4 训练集规模的讨论
使用箱线图来评估不同训练集规模在本文模型上的结果,箱线图不仅可以显示精度在不同训练集大小下的变化,而且可以显示每个实验的稳定性.从图4b可以看出,本文模型的效果很大程度上受到训练数据集大小的影响,准确率也随着训练样本数量的减少而下降.此外,NSJL模型在每个训练集规模下的性能都是相对稳定的.综上所述,训练数据集的大小对多阶段疟原虫识别任务起着重要的作用.
3.4.5 参数分析
在此,用不同的参数k和学习率来评价NSJL模型的性能.k和学习率不同时准确率的结果分别如图4c和4d所示.较大的k可能导致异类间样本相连,而较小的k可能导致建图过程中同类样本无法有效连接,因此适当的k值可以达到最好的结果,如图4c.类似地,如图4d,更大的学习率容易将模型陷入局部最优解而较小的学习率将导致收敛速度变慢.
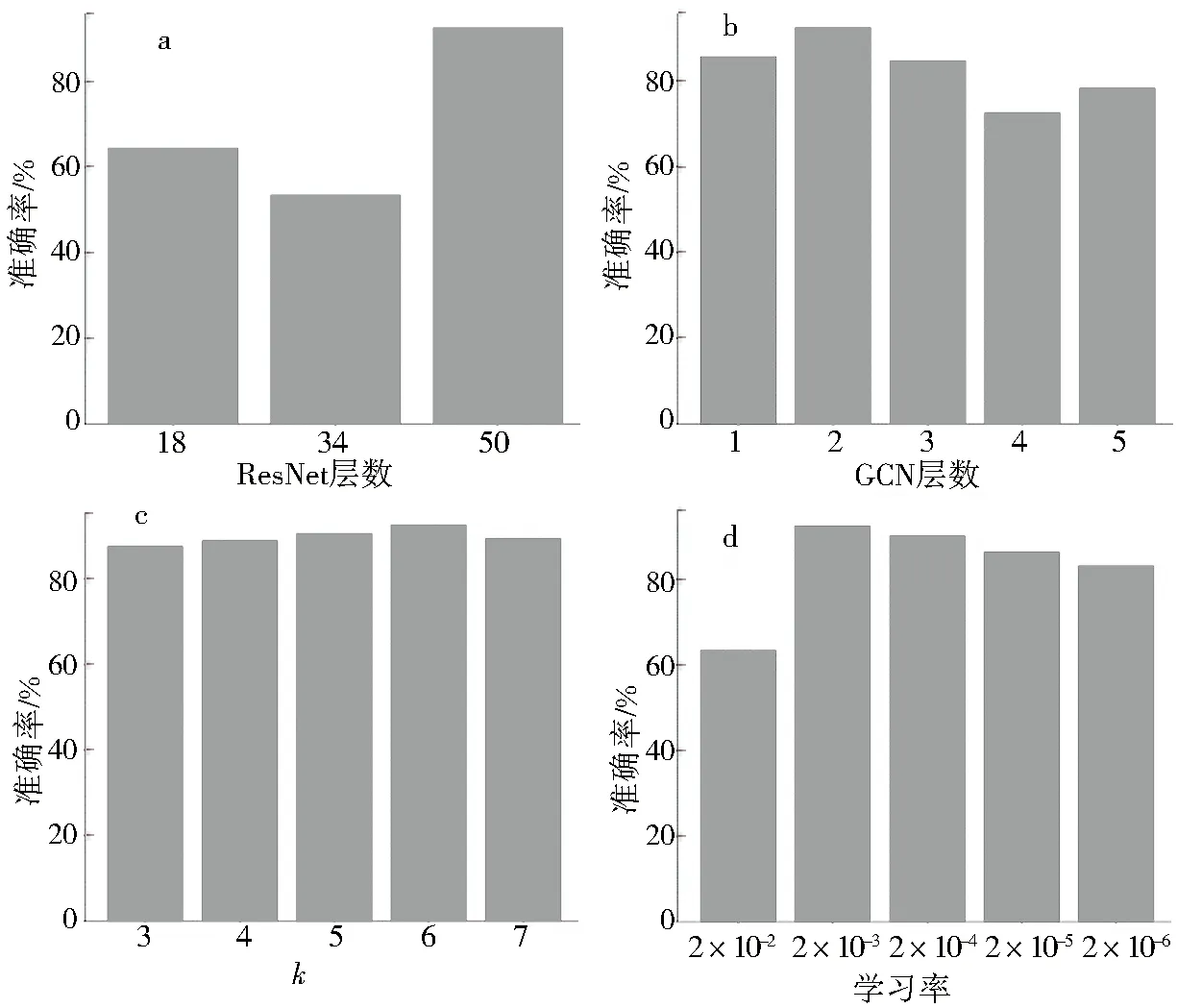
a.ResNet;b.GCN;c.K-NN;d.学习率.图4 当网络参数和网络深度发生变化时,NSJL模型的准确率Fig.4 Accuracy results of NSJL model when parameters and depth of networks are changed
4 结论
本文提出了近邻样本联合学习(neighbor sample joint learning,NSJL)模型来解决多阶段疟疾识别问题.NSJL将CNN特征学习、邻域相关性挖掘和图嵌入学习模块集成到一个统一的框架中,用于判断每个细胞的感染阶段.
在NSJL模型出现之前,基于深度学习的计算机疟疾自动识别模型都集中于二分类问题,即只判断待识别细胞是否被疟疾寄生虫感染,但无法判断细胞被感染的阶段.NSJL模型首次完成了疟疾多阶段识别的任务,不仅能够从显微图像中识别出正常的红细胞和白细胞,还能准确区分出异常细胞中细胞被疟疾感染的程度,从而给医生提供了更准确更全面的识别结果,进一步加快了医生的就诊效率.除此之外,现有的疟疾识别模型都是基于CNN特征提取,虽然能学习单个图像的特征,却无法得到图像间的关系,从而丢失了大量的关键信息.NSJL通过邻域相关性挖掘模块提取不同疟疾样本间的关系,并通过图嵌入学习模块使用样本间关系和CNN特征共同进行疟疾识别,极大地提高了疟疾识别的准确率.NSJL模型能够快速、准确、详细地将识别结果提供给相关医生,促进了疟疾自动识别模型的推广.通过实验验证了该模型相对于3种公认的网络的优越性,也表明该模型能够有效地解决多阶段疟疾识别任务.
但是,NSJL模型也存在着不足,深度学习方法需要大量提前标注的真实标签作为训练依据,标签的标注工作也需要占用大量经验丰富的专家的精力.而如果真实标签过少或者存在较大的噪声,则会严重影响模型的疟疾识别能力.因此对真实标签的需求是NSJL模型的主要不足.后续将在该模型中引入半监督或迁移学习的思想,研究模型从无标签数据中学习关键信息或通过引入带有标签的外部数据来解决这个问题.
猜你喜欢
临床内科杂志(2022年4期)2022-11-24
农业工程学报(2022年12期)2022-09-09
计算技术与自动化(2022年1期)2022-04-15
北京航空航天大学学报(2021年9期)2021-11-02
北京航空航天大学学报(2021年9期)2021-11-02
疯狂英语·新阅版(2020年4期)2020-05-29
环球时报(2019-09-11)2019-09-11
领导决策信息(2018年16期)2018-09-27
人大建设(2017年10期)2018-01-23
数学学习与研究(2017年3期)2017-03-09
