
宏基因组技术鉴定红原县儿童腹泻相关病毒
2022-04-20 07:10杨贵生
西南民族大学学报(自然科学版) 2022年2期
宋 哲,敬 琼,杨贵生,周 燕,张 斌,汤 承,岳 华
(1. 西南民族大学畜牧兽医学院,四川 成都 610041;2. 四川省阿坝藏族羌族自治州疾病预防控制中心,四川 马尔康 624000)
腹泻是我国儿童最常见的一种疾病,是影响儿童健康的主要公共卫生问题.病毒是引起儿童腹泻的主要的原因[1-2],其中轮状病毒(Rotavirouses,RV)、诺如病毒(Norovirus,NoV)、札幌病毒(Sapovirus,SV)、肠道腺病毒(Enteric Adenoviruses,EAd)、星状病毒(Astroviruses,AstV)等最为常见[3].不同地区导致儿童腹泻的病毒种类和流行情况存在一定的差异[3-5],而我国经济不发达地区相比发达地区的儿童腹泻患病率要更高[6-7].有研究资料显示西藏、四川、宁夏等地少数民族聚居区儿童腹泻远高于全国平均水平,可能与地区经济、生活习惯和卫生资源分布存在的差异性等有关[8-9].红原县位于四川省阿坝藏族羌族自治州,地处青藏高原东南沿,是典型的高原牧区,儿童腹泻是当地的常发病和多发病,但儿童腹泻相关的病原学和流行病学资料严重匮乏,制约了当地儿童腹泻病的诊断和防控.
病毒宏基因组技术具有能够直接从样本中无偏差鉴定病毒种类,发现新病毒,了解病毒的遗传多样性和较为容易的获得病毒基因组等优势[10].2011 年单同领等采用此技术,分析了上海腹泻儿童粪便中病毒的群落,为儿童腹泻病毒的诊断提供了参考[11];2012 年Phan 等从西非的儿童腹泻粪便中鉴定到多种致腹泻病毒,并发现了一种新的 bufavirus 病毒[12];2017 年赵小英等在我国江苏的儿童腹泻粪便中鉴定出多种致腹泻病毒,并获得了一株新型人双埃可病毒基因组,在此基础上对该新型病毒的分子流行病学进行深入研究[13];这些研究体现出了病毒宏基因组技术在儿童腹泻病毒鉴定的优势.本研究目的是调查红原县儿童腹泻粪便中的病毒种类及其分子特征,为当地儿童腹泻的诊断和防控提供参考,丰富青藏高原儿童腹泻病毒的遗传进化资料.
1 材料和方法
1.1 样品采集
12 份粪便样本来自2019 年5 ~11 月红原县 4 月龄~11 岁急性胃肠炎患者.
1.2 试剂
QIAamp Viral RNA Mini kit 购自德国 QIAGEN 公司;Maxima H Minus Doubles-Stranded cDNA Synthesis Kit 购自中国Thermo 公司;PMD19-T 载体购自宝生物工程 (大连) 有限公司;大肠杆菌DH5α 感受态细胞、DNase 酶、RNase 酶、DNA Marker、DNA 纯化试剂盒和Plasmid Miniprep Kit 均购自美国AXYGEN 公司.
1.3 核酸的提取、cDNA 的合成及测序
样本于冰上解冻,涡旋20 s 混匀,5 000 r/min 离心5 min,上清液用0.22 μM 滤器过滤,加入2%的双抗,样本于-80 ℃冻融 3 次,采用 QIAamp Viral RNA Mini kit 试剂盒分别提取12 份样本的核酸.5 ~6 月的6 份样本组成一个 pool 样,10 ~ 11 月的 6 份样本组成一个 pool 样,加入 10 U 的 DNase 和 1.5 μg RNase 在37 ℃条件下孵育90 min 以除去游离的核酸. 按照Thermo Scientific Maxima H Minus Double-Stranded cDNA Synthesis Kit 反转录试剂盒和随机引物的说明书合成双链cDNA,通过Qubit 分光光度计测定pool 样中cDNA 的质量,检验合格的cDNA 送派森诺生物,利用Illmina 公司的NovaSeq 二代测序平台对两个文库进行测序,使用MEGAN 软件分析病毒种类.
1.4 基因组的验证
根据来自Illumina 测序得到的病毒序列,设计病毒的检测引物并建立检测所鉴定病毒的RT-PCR 方法,以进一步验证病毒宏基因组鉴定结果的准确性,并调查各种病毒在各样本中的分布,引物信息见表1.反应体系:酶12.5 μL,上下游引物(10 μmol/L)各0.4 μL,DNA 或 c DNA 2 μL,ddH2O 补足25 μL.反应条件:95 ℃预变性5 min;95 ℃变性30 s,退火温度见表1,72 ℃ 延伸 1 min,共 40 个循环;72 ℃ 延伸10 min.PCR 产物经1.5%琼脂糖凝胶电泳鉴定,用胶回收试剂盒回收目的片段,并将其克隆至pMD19-T载体,并转化大肠杆菌DH5α 感受态细胞,筛选出阳性克隆接入含氨苄青霉素的LB 液体培养基中,37 ℃培养8 h,用质粒提取试剂盒提取重组质粒,送擎科生物有限公司测序,采用DNAMAN 软件进行序列分析.

表1 检测6 种病毒的引物信息Table 1 Primer information for detection of 6 viruses
1.5 AstV、NoV、SV 基因组扩增
为了进一步了解AstV、NoV、SV 的基因组特征,根据本研究获得的数据和 GenBank 收录的 AstV、NoV、SV 毒株序列,利用Primer 5.0 分别设计引物,引物相关信息如表2 所示.

表2 扩增基因组的各引物信息Table 2 Primer information of amplified genome
1.6 AstV、SV、NoV 的基因组序列和遗传进化及重组分析
将获得的各目的片段利用DNAstar 中的Seqman进行拼接; 用 ORF 查找软件(http:/ /www. ncbi.nlm. nih. gov/gorf/gorf. html)进行分析;使用DNAStar软件中的MegAlign 程序分析核苷酸和氨基酸序列的相似性.使用MEGA7.0 软件以Neighbor-joining(Bootstrap 值为1 000)构建系统进化树对获得的病毒基因组进行遗传进化分析. 用 SimPlot 软件(Version 3.5.1)和Recombination Detection Program 4.0(RDP 4.0,Version 4.96)的 RDP,Gene Conv,Chimaera,Max-Chi,BootScan,Si Scan 和 3Seq 方法预测重组事件.
2 结果
2.1 腹泻粪便样本中病毒种类
两个文库共获得318710620 reads,可注释为病毒序列的读长为1590815 reads ,并拼接出2 898 条病毒contig.经过序列比对后,从红原县儿童腹泻粪便中共鉴定出6 种病毒:AstV、AdV、NoV、SV、CAV 和 AiV,其序列组成见图1.
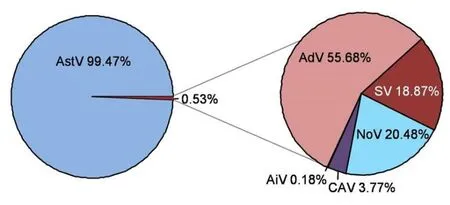
图1 腹泻儿童粪便样本中病毒的序列组成Fig.1 Sequence composition of viruses in fecal samples
2.2 病毒在12 份样本中的分布
RT-PCR 证实了6 种病毒的真实存在,检出率分别为:AstV:66.67%、NoV:8.33%、SV:25.00%、AiV:8.33%、CAV:8.33%、AdV:8.33%. 混合感染检出率为33.33%.其在各样本中的分布见表3.
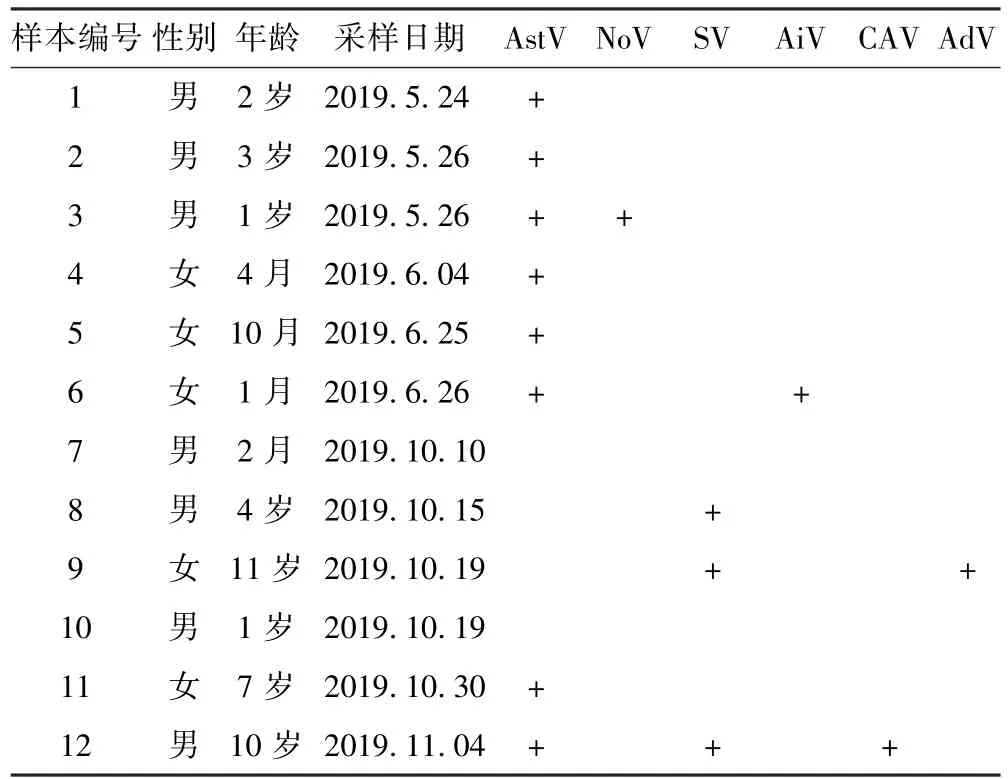
表3 6 种病毒在临床样本中的分布Table 3 Distribution of 6 viruses in clinical samples
2.3 AstV 基因组分析
成功从2 号样本中获得一条AstV 基因组,命名为Hu/HY/2019/CHN(GenBank 登录号:MW863309).完整基因组全长6 826 bp,编码框全长6 645 bp,GC含量 44.37%,由 5′UTR 和 ORF1a、ORF1b、ORF2 以及 3′UTR 组成,5′UTR 为 76 bp,ORF1a 为 2 803 bp,编码934 个氨基酸,ORF1b 为 1 561 bp,编码 520 个氨基酸,ORF2 为 2 364 bp,编码 788 个氨基酸,3′UTR为81 bp. 该毒株与GenBank 中上海人源 AstV 毒株SH1(FJ357759)相似性最高(97.3%),为 AstV-1b型.与 SH1 序列比对发现,5′UTR 存在 5 个碱基的突变,3′UTR 相有一个碱基的突变,ORF1a 和 ORF1b 共有96 个碱基突变(30 个氨基酸突变),ORF2 有 43 个碱基突变(5 个氨基酸突变). 经预测抗原表位后,发现氨基酸突变并未处于抗原表位位置.
为了进一步分析Hu/HY/2019/CHN 的进化关系,选取了GenBank 中国内所有的AstV 基因组序列、11 个与其相似性最高的国外毒株基因组序列和其它型代表毒株基因组序列,采用邻接法构建进化树,结果显示与上海株SH1(FJ375759)遗传关系最近(图2).基于ORF1a、ORF1b、ORF2 分别建立的进化树也与上海株SH1(FJ375759)遗传关系最近(资料未显示).
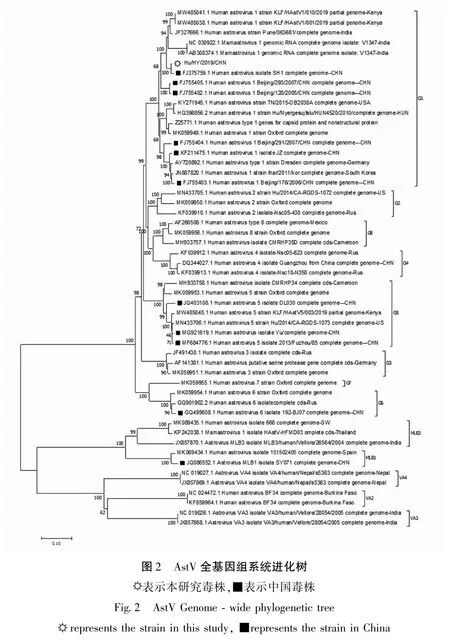
2.4 SV 基因组分析
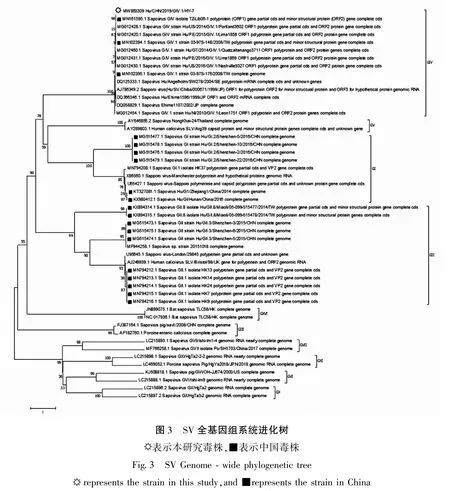
成功从9 号样本中获得一条片段为7 355 bp ,其中编码框全长为7 302 bp 的SV 基因组,命名为Hu/CHN/2019/GIV.1/HY-7(GenBank 登录号:MW86330 9),GC 含量为 53.23%.由 5′ UTR 和 ORF1、ORF2 及3′UTR 组成.5′UTR 长度为 16 bp,ORF1 与 ORF2 之间有4 个核苷酸的重叠.ORF1 的长度为6 803 bp,编码2 266 个氨基酸,依次为 NS1、NS2、NS3、NS4、NS5、NS6-7 和 VP1;ORF2 长度为 504 bp,编码 168 个氨基酸,构成 VP2 蛋白.3′UTR 长度为 36 bp. 与 GenBank中江苏毒株TZ-Lib06-1(GenBank 登录号:MN16159 5)相似性最高(98.47%),为 GIV.1 型. 与 GenBank中所有的34 条完整GIV.1 型 SV 相比,ORF1 区域有43 个独特的核苷酸的突变, 9 个核苷酸突变位于RdRp区域,A1304T 核苷酸突变为有义突变使得RdRp基因编码的氨基酸发生了独特变异(N435T);15 个核苷酸突变位于VP1 区域,A173T 核苷酸突变为有义突变,使得VP1 基因编码的氨基酸发生了独特的变异(V58E);VP2 区域有两个独特的核苷酸突变,未见有义突变.
选择GenBank 中的全部国内SV 毒株的基因组序列、10 个与其相似性最高的国外毒株基因组序列和其它型代表毒株的基因组序列采用邻接法构建进化树(图 3),Hu/CHN/2019/GIV.1/HY-7 与江苏株MN161595 遗传关系最近. 基于 SV 的 VP1、VP2 和RdRp 区氨基酸序列构建进化树,结果与全基因组系统发育分析结果一致(资料未显示).
2.5 NoV 基因组分析
成功从3 号样本中获得一条片段为7 554 bp,其中编码框全长为6 702 bp 的NoV 基因组,命名为Hu/HY-1/19/CHN(GenBank 登录号:MW981327),GC 含量为 49.85%. 由 5′UTR 和 ORF1、ORF2、ORF3 以及3′UTR 组成.5′UTR 为5bp, ORF1 的长度为5 097 bp,依次编码 p48、NTPase、p22、Vpg、 Pro、RdRp;ORF2 的长度为 1 623 bp,编码 VP1 蛋白;ORF3 长度为8 076 bp,编码 VP2 蛋白,3′UTR 为41 bp.使用诺如病毒在线分型网站进行分型,结果为GII.4 型Sydney-2012 类基因簇,与我国河南毒株MK934772 相似性最高,核苷酸相似性为98.73%,氨基酸相似性为97.1%.
与GenBank 中相似性最高的 100 条 GII.4 型Sydney-2012 类基因簇毒株的 RdRp 区域相比,Hu/HY-1/19/CHN 有23 个独特的核苷酸突变(A126G、A147G、C159T、T175C、G177A、A222G、C267T、C303T、G366A、T418C、T453C、G546A、T705C、T729C、A820G、C978T、 C979T、 C993T、 T996C、 G1101A、 C1110T、A1271G、C1365T),其中 A820G 和 A1271G 位置的核苷酸位点为有义突变导致了氨基酸位点的突变(I274V、E424G),分别位于 RdRp 手掌和手指结构域.与GenBank 中相似性最高的100 条GII.4 型Sydney-2012 类基因簇毒株的VP1 区域的抗原表位A、B、B2、D、E、F 进行位点突变的分析,结果显示在抗原表位F 中有1 个独特突变(D357G),位于P2 结构域.
出于篇幅考虑,只选择了GenBank 中与Hu/HY-1/19/CHN 相似性最高的 30 条(相似性 97.73%-98.73%)全基因组序列进行系统发育分析,所有毒株均为 GII.4 型 Sydney-2012 类基因簇,Hu/HY-1/19/CHN 聚为独立一支,显示出独特的进化趋势(图4).基于VP1 蛋白和RdRp 蛋白的氨基酸序列分别建立的进化树,结果均与Sydney-2012 类基因簇毒株聚为一支,分别与广东毒株(KY407116) 和济南毒株(MG214988)遗传关系最近(资料未显示).

3 讨论
四川省阿坝藏族羌族自治州红原县属于典型的高原牧区,儿童腹泻是当地的常发病和多发病,但儿童腹泻相关的病原学和流行病学资料严重匮乏.本实验采用了宏病毒组技术从红原县12 份儿童腹泻粪便中鉴定出6 种致腹泻病毒(AstV、NoV、SV、AiV、CAV、AdV),并用RT-PCR 进一步证实了这6 种病毒的存在,其中星状病毒的检出率最高.12 份样本中有6 份单一病毒感染,4 份样本为病毒混合感染. 病毒的混合感染会增加疾病的严重性,同时也增加了诊断和防控的难度[14].本实验结果为当地儿童腹泻的病原学诊断提供了参考,也为进一步开展腹泻病毒的分子流行病学的调查奠定了基础.
AstV 是导致婴幼儿腹泻的重要病原之一[15]. 在本实验的12 份样本中,AstV 检出率为66.67%,春季和秋季的样本中均有检出. 因此,AstV 可能是导致当地儿童腹泻的一个重要原因. 本实验AstV 的检出率显著高于2017 ~2019 年四川地区1.89% 的AstV 检出率[16],这可能与采样地点不同有关. 目前在Gen-Bank 中中国AstV-1b 型完整基因组共5 条,包括本研究上传的Hu/HY/2019/CHN(MW863309)毒株,采样年份分别为 2005、2007、2008、2010、2019 年. AstV 基因ORF2 中的两个可变区VR1 和VR2 被用来评估HAstV 的变异,将5 条序列的 VR1 和 VR2 氨基酸序列进行比对,结果显示差异不大,提示我国AstV-1b型的序列仍相对稳定,这与Guo 的研究结果一致[17].
在本实验采集的12 份样本中 SV 检出率为25.00%,均为秋季采集的样本,这可能与SV 以寒冷季节较为流行有关[18].2001 年我国首次报道了SV 的检出[19],该病毒已经在国内广泛流行,检出率在0.70% ~2.50%[20-22].2017 ~2019 年在四川 1 ~50 岁人群中709 份样本SV 检出阳性率为2.50%[16],低于本实验在红原县样本的检出率,这可能和样本的时间和采样的地点有一定关系.本研究从腹泻粪便样本中获得了1 株SV 基因组序列,为GIV.1 型.SV 的RdRp与基因组的复制以及亚基因组RNA 的合成和扩增中起关 键 作 用[23], 值 得 注意 的是 Hu/CHN/2019/GIV.1/HY-7 与其它GIV.1 型SV 的RdRp 区域相比,有9 个独特的核苷酸突变,其中有1 个核苷酸突变为有义突变,使得435 位氨基酸由N 变为T,此独特变异是否与青藏高原地区独特的地理环境有关以及对功能的影响有待进一步研究.
NoV 是全球流行的急性胃肠炎病因之一[24]. 本实验12 份样本中检出了一份阳性,并成功获得了NoV 基因组序列,为 GII.4 型 Sydney-2012 簇成员.遗传进化分析Hu/HY-1/19/CHN 聚为独立一支,显示出独特的进化趋势. GII.4 型Sydney-2012 株于2013年由澳大利亚学者首次报道[25],此后在世界范围内流行[26-27];2013 年国内也证实了该型毒株的存在,此后在广东、江苏、四川、北京、安徽等地陆续报道,也成为了我国的流行毒株[28-30].抗原变异是导致新型NoV出现的重要因素之一[31],Sydney-2012 的VP1 蛋白中的368、373 和376 位点的适应性变化由GII.4-2009产生抗原变异而出现,与其它型的基因簇相比,Sydney-2012 能够结合更广谱的 HBGAs,因此导致其广泛的流行[32]. 本研究中的Hu/HY-1/19/CHN 与其它GII.4 型 Sydney-2012 毒株的 VP1 区域相比,其 P2 结构域F 抗原表位中有1 个独特突变(D357G),而表位F 是目前已知的唯一保守的GII.4 封锁表位,在1974~2015 年GII.4 株中均为保守的,F 表位的氨基酸突变,可能会导致免疫逃逸变异[33],因此,有必要进一步监测Hu/HY-1/19/CHN 毒株的流行性情况.
NoV 的RdRp 对病毒的复制十分重要,大流行性GII.4 型人源诺如病毒RdRp 的低保真度有助于其成为优势毒株全球流行[33]. 与其它 GII.4 型 Sydney-2012 毒株的 RdRp 区域相比,Hu/HY-1/19/CHN 有 2个氨基酸位点的突变(I274V、E424G),分别位于RdRp 手掌和手指结构域,是与病毒的复制与合成密切相关的结构域[34],因此氨基酸的突变对RdRp 功能的影响值得进一步研究.
4 结论
本研究首次在红原县儿童腹泻样本中证实AstV、SV、NoV、AiV、CAV、AdV 6 种病毒的存在,AstV 的检出率最高,存在病毒混合感染的情况. 成功获得了AstV-1b 型、GII.4 型 NoV 和 GIV.1 型 SV 毒株的基因组.本实验结果为红原县儿童腹泻的病原学诊断和流行病学调查提供了参考,丰富了青藏高原儿童腹泻病毒的分子特征和遗传进化资料.
猜你喜欢
动物医学进展(2022年9期)2022-11-26
肝博士(2022年3期)2022-06-30
昆明医科大学学报(2022年4期)2022-05-23
中国药学药品知识仓库(2022年1期)2022-03-23
科学大观园(2022年2期)2022-01-23
中国体育教练员(2021年4期)2022-01-21
文萃报·周二版(2021年47期)2021-12-14
烹调知识(2019年3期)2019-03-01
中国纤检(2015年8期)2015-05-08
