
基于ReaxFF MD方法的煤焦增压富氧燃烧模拟
2022-04-18 08:05洪迪昆翟晓明
动力工程学报 2022年4期
洪迪昆,雷 鸣,翟晓明,郭 欣
(1.华北电力大学 动力工程系,河北保定 071003;2.华中科技大学 煤燃烧国家重点实验室,武汉 430074)
燃煤会排放大量CO2等温室气体,这是造成全球气候变暖的主要原因之一,因此实现燃煤过程中CO2的捕获迫在眉睫[1]。富氧燃烧技术被认为是燃煤电站最具应用前景的CO2捕获技术之一。该技术不仅可以实现烟气中CO2的富集,还能降低NOx、SOx等污染物的排放。然而,在常规富氧燃烧技术中空气分离与CO2压缩过程均在高压下进行,而富氧燃烧却在常压下进行,由于系统经历升压、降压、再升压过程,导致能量损失严重[2]。近年来,增压富氧燃烧技术越来越受到关注。由于高压下水蒸气凝结温度升高,烟气中难以回收的潜热可以用于凝结换热,弥补了维持系统高压运行所损失的循环效率;而且由于高压设备结构紧凑且规模较小,可在一定程度上节省电站基建投资[3];此外,由于燃烧在高压下进行,只需将锅炉排烟冷却到20 ℃以下,无需压缩烟气便可直接得到液态CO2,节约了系统能耗。由此可见,增压富氧燃烧技术将是未来燃煤电站CO2捕获的主要方向之一。
已有学者对不同压力下煤的富氧燃烧特性进行了研究。雷鸣等[2]在加压热天平上研究了烟煤在增压富氧条件下(0.1~3 MPa)的燃烧行为,结果显示挥发分的燃烧速率随着压力的升高逐渐增大。Joutsenoja等[4]在加压流化床上研究了0.2~1 MPa压力范围内褐煤的富氧燃烧特性,发现压力对燃烧速率的影响较小。盛金贵等[5]采用理论研究的方法探索了增压富氧条件下压力(0.1~2.5 MPa)对煤焦燃烧速率的影响,结果表明增压显著提高了煤焦的燃烧速率,当压力升高到一定值后,再加压对煤焦燃烧速率的影响逐渐减小。尽管这些研究在一定程度上揭示了压力对富氧燃烧的影响规律,然而由于实验条件的限制,大多数研究在0.1~3 MPa压力范围内进行,无法体现实际工况条件下(4.83~8.96 MPa)的富氧燃烧特性。此外,目前对增压富氧燃烧的实验研究主要在高压热重分析仪、高压固定床和高压流化床上进行,侧重于总包反应动力参数的获得,难以揭示压力对富氧燃烧的影响机制。实际上,压力对煤焦氧化反应速率和气化反应速率均有显著的影响[6]。因此,如何全面系统地定量分析不同压力下CO2气化反应对煤焦转化的净贡献仍有待进一步研究。
基于反应力场的分子动力学(ReaxFF MD)方法[7]被广泛应用于燃烧领域。Cheng等[8-9]采用ReaxFF MD方法研究了甲苯和正十二烷的燃烧机理,考察了温度和压力等条件对燃烧的影响,建立了不同压力条件下的燃烧动力学模型。Castro-Marcano等[10]通过ReaxFF MD方法研究了不同条件下Illinois 6号煤焦的燃烧反应,发现煤焦的燃烧反应是由煤焦分子的热分解所激发的,随后再发生氧化反应,也有部分煤焦与氧气发生攫氢反应生成H自由基和OH自由基。这些研究显示了ReaxFF MD方法在燃烧反应机理研究方面的独特优势,但均只针对燃料/O2两组分体系进行研究,无法体现富氧燃烧过程中CO2的气化作用。Hong等[11-12]将ReaxFF MD方法应用于CH4/O2/CO2/H2O多组分体系燃烧的研究中,探索了CO2与 H2O的化学作用对 CH4燃烧的影响机理,揭示了CO2与H2O之间的协同/竞争机制。
笔者以准东五彩湾煤焦为研究对象,基于实验表征结果构建其分子结构模型,采用ReaxFF MD方法在4~10 MPa压力范围内对煤焦的富氧燃烧进行模拟,探索压力对氧化反应和CO2气化反应净贡献的影响规律,建立不同压力下煤焦氧化反应和CO2气化反应的动力学模型,并通过分析煤焦结构的演变从分子层面上揭示不同压力下煤焦富氧燃烧的微观机理。
1 模拟方法
1.1 煤焦分子结构模型
构建合理的煤焦分子结构模型是采用ReaxFF MD方法研究煤焦富氧燃烧机理的基础。对准东五彩湾煤焦进行元素分析,结果如表1所示,其中nHC和nOC分别为煤焦中H、C物质的量比和O、C物质的量比。

表1 准东五彩湾煤焦元素分析Tab.1 Ultimate analysis of Wucaiwan coal char


图1 准东五彩湾煤焦XPS 谱图及分峰拟合Fig.1 XPS spectrogram and multi-peak fitting of Wucaiwan coal char

图2 准东五彩湾煤焦分子结构模型Fig.2 Molecular structure model of Wucaiwan coal char
1.2 反应力场
量子化学方法可以精确描述化学反应过程,但是仅针对较小体系且不能体现温度、压力等条件对反应的影响;传统的分子动力学方法可以从分子层面对复杂体系的能量、结构和振动加以描述,但是不能描述体系的化学反应过程。基于反应力场(ReaxFF)的分子动力学方法结合了量子化学方法和传统分子动力学方法的优点,可以描述较大体系的化学反应过程且能体现温度、压力的影响。反应力场通过原子间瞬时距离的键级来计算键能、键角能和二面角能,并利用键断裂和形成过程中键级的变化来描述真实结构模型中的化学反应,而键级是根据原子之间的距离计算而得来的,考虑了单键、π键和ππ键。同时,反应力场也能描述范德华相互作用及库伦相互作用。在反应力场中,系统的能量Esystem为:
(1)
式中:Ebond为键结能;Eover、Eunder、Eval、Epen、Etors、Econj、EvdW及ECoul分别为过饱和键能、不饱和键能、键角能、共价键修正能、二面角能、共轭能、范德华相互作用力能及静电相互作用能。
1.3 模拟过程
首先,使用Materials Studio软件中的Amorphous Cell模块构建煤焦/O2/CO2体系模型(包含1个煤焦分子、135个O2分子和500个CO2分子),体系的初始密度设为0.01 g/cm3。其次,对模型进行能量最小化计算,使体系的能量以及各原子的位置坐标更加合理,避免因原子的重叠而无法进行动力学平衡计算。随后,分别在4 MPa、7 MPa和10 MPa压力下进行1 000 ps(1 ps=10-12s)的NPT(其中系统的粒子数N、压力p和温度T恒定)动力学平衡计算。模拟过程采用周期性边界条件[13],时间步长设为1 fs(1 fs=10-15s),运动方程数值积分采用Verlet算法,体系的温度和压力控制均采用Berendsen方法[14]。图3给出了不同压力下NPT动力学平衡计算时体系密度随时间的变化。由图3可以看出,4 MPa、7 MPa和10 MPa压力对应的体系最终密度分别为0.061 g/cm3、0.127 g/cm3和0.208 g/cm3。最后,采用Lammps软件对不同压力下的体系进行ReaxFF MD模拟。体系温度从2 000 K升至4 000 K,升温速率为2 K/ps,系综选取NVT(其中系统的粒子数N、体积V和温度T恒定)。模拟的时间步长为0.1 fs,截断值为0.3。ReaxFF选取C/H/O/N力场参数。值得注意的是,ReaxFF MD模拟温度(2 000~4 000 K)比实验温度(通常为1 000~2 000 K)高,这是因为模拟在较短的时间内进行(皮秒级别),远远低于实验反应时间(秒级别),因此提高模拟温度可增加原子间的碰撞,从而使得反应能够在有限的时间内发生。虽然模拟温度与实验温度存在差异,但是已有研究[15]证明了ReaxFF MD模拟中提高温度策略的合理性。

图3 不同压力下NPT优化时体系密度随时间的变化Fig.3 Change of system density with time during NPT optimization at different pressures
2 结果与分析
2.1 压力对煤焦碳转化率的影响
图4给出了不同压力下富氧燃烧过程中煤焦整体碳转化率随反应时间的变化曲线。由图4可以看出,在0~600 ps时间内(对应温度为2 000~3 200 K)4 MPa和7 MPa压力下的碳转化率基本相当,而10 MPa压力下碳转化率增加;在>600~1 000 ps时间内(对应温度为>3 200~4 000 K)碳转化率随着压力的升高而增加。在4 MPa压力下,煤焦的最终碳转化率为86.57%;在7 MPa和10 MPa压力下,煤焦分别在950 ps和850 ps转化完全。
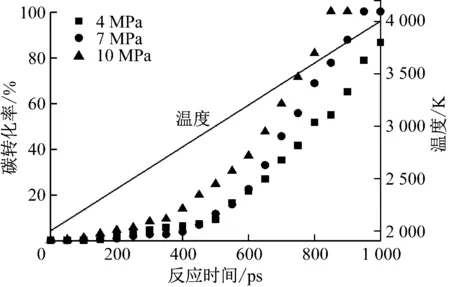
图4 不同压力下富氧燃烧过程中煤焦碳转化率随反应时间的变化Fig.4 Change of carbon conversion of char with reaction time during oxy-fuel combustion at different pressures
富氧燃烧过程中煤焦的消耗主要发生在其氧化反应和气化反应中。图5给出了不同压力下体系中O2和CO2分子数量随反应时间的变化曲线。由图5(a)可以看出,在0~600 ps时间内O2分子消耗量在4 MPa和7 MPa压力下基本相当,而在10 MPa压力下O2分子的消耗量明显较多;在>600~1 000 ps时间内O2分子消耗量随着压力的升高而增加,高压促进了煤焦的氧化反应。由图5(b)可以看出,在0~600 ps时间内CO2分子消耗量在4 MPa和7 MPa压力下基本相当,而在10 MPa压力下CO2分子几乎无消耗;在>600~1 000 ps时间内CO2分子消耗量随着压力的升高而减少,表明高压抑制了煤焦的气化反应。

(a)O2分子
2.2 氧化反应和气化反应对煤焦消耗的净贡献


图6 不同压力下煤焦碳转化率计算值与真实值的对比Fig.6 Carbon conversion comparison of char between calculated values and actual values at different pressures
(2)
(3)
χtotal=χoxidation+χgasification
(4)
式中:Ninitial-CO2为初始CO2分子数量,本文中取500;NCO2为反应过程中CO2分子数量;N0为煤焦中初始碳原子的数量,本文取268;Ninitial-O2为初始O2分子数量,本文取135;NO2、NH2O、NOH及NO分别为反应过程中O2分子、H2O分子、OH自由基和O自由基的数量。
基于式(2)和式(3),分别计算得出气化反应和氧化反应对煤焦转化的净贡献,结果如图7所示。由图7(a)可以看出,氧化反应对煤焦转化的净贡献随着压力的升高而增加,4 MPa、7 MPa和10 MPa压力下最终的氧化反应对煤焦转化的净贡献分别为39.29%、58.86%和75.59%。由图7(b)可以看出,气化反应对煤焦转化的净贡献随着压力的升高而降低,4 MPa、7 MPa和10 MPa压力下最终的气化反应对煤焦转化的净贡献分别为47.28%、40.90%和23.78%。在4 MPa和7 MPa压力下,气化反应的煤焦转化净贡献随着反应时间的增加不断增加;而在10 MPa压力下气化反应的煤焦转化净贡献在接近700 ps时才开始不断增加,且在850 ps后保持恒定,这是因为850 ps时煤焦几乎被消耗完全。
为了进一步揭示压力对煤焦氧化反应和气化反应的影响机制,对不同压力下的O2和CO2扩散速率进行了分析。图8给出了不同压力下O2和CO2分子的均方根位移(MSD)曲线。气体分子的扩散系数可通过MSD进行计算:
(5)
式中:D为气体分子的扩散系数;r(0)为分子的初始位置坐标;r(t)为分子在t时刻的位置坐标;n为体系中分子或原子总数量。
因此,可以通过MSD曲线的斜率来判断分子的扩散性。由图8可以看出,O2和CO2分子的扩散系数均随着压力的升高而降低。对于氧化反应而言,虽然增压降低了O2的扩散系数,但是提高了煤焦周围的O2浓度,从而提高了煤焦的氧化反应速率。对于气化反应而言,燃烧体系中CO2的体积分数高达80%,增压对提高煤焦周围CO2体积分数的作用并不显著,CO2的扩散对气化反应的影响更加显著,因此高压降低了煤焦的气化反应速率。另外,在富氧燃烧过程中O2与CO2会竞争煤焦的活性碳位点,随着压力的升高,CO2与O2竞争活性碳位点的能力可能减弱,从而导致高压下煤焦-CO2气化反应净贡献降低。
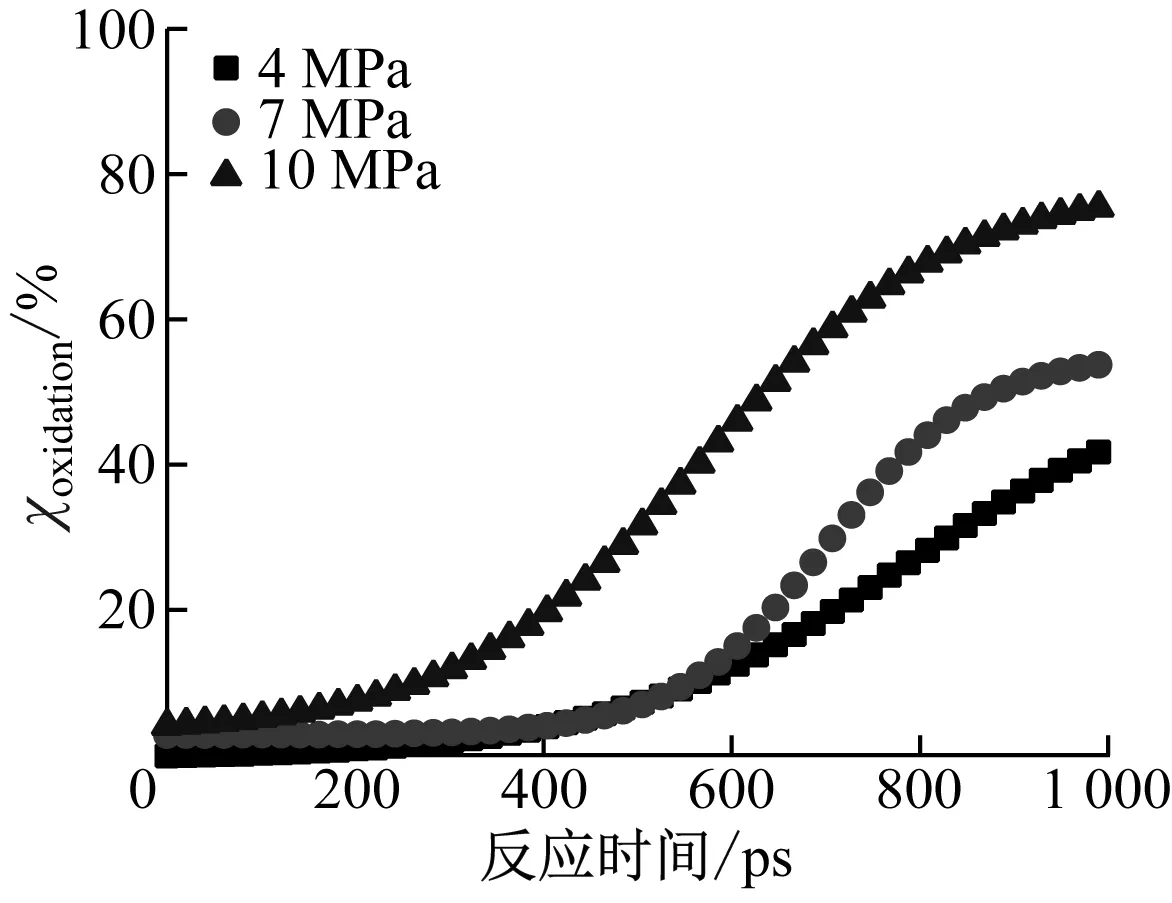
(a)氧化反应

(a)O2分子
2.3 压力对煤焦氧化反应和气化反应动力学的影响
为进一步分析压力对煤焦氧化反应和气化反应动力学的影响,基于图7中的煤焦碳转化率从动力学层面去定量表征不同压力下的反应过程。采用非等温一级反应动力学模型,如式(6)所示。
(6)
式中:K为反应速率;X为煤焦碳转化率;A为指前因子;R为气体常数;β为升温速率;Ea为反应活化能。
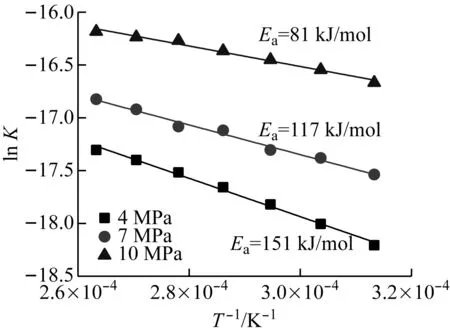
图9给出了不同压力下煤焦氧化反应和气化反应动力学拟合曲线。由图9(a)可以看出,4 MPa、7 MPa和10 MPa压力下煤焦氧化反应的活化能分别为151 kJ/mol、117 kJ/mol和81 kJ/mol,表明增压能够降低煤焦氧化反应的活化能,这可能是因为增压降低了O2的扩散系数,在较高压力下煤焦的氧化反应更易受扩散控制,从而具有较低的活化能。由图9(b)可知,4 MPa、7 MPa和10 MPa压力下煤焦气化反应的活化能分别为174 kJ/mol、166 kJ/mol和172 kJ/mol(由于10 MPa压力下气化反应在700 ps才开始发生,且850 ps后煤焦基本消耗完全,因此10 MPa压力下的动力学拟合温度区间较窄),表明压力对煤焦气化反应活化能的影响并不明显。
2.4 压力对煤焦结构的影响
为了探究压力对煤焦结构演变的影响,对模拟轨迹文件进行分析。不同压力下煤焦结构的演变如图10所示。在4 MPa压力下2 000 K、3 000 K、3 200 K、3 400 K、3 600 K和 3 800 K温度对应的煤焦分子式分别为C268H30O15N2、C244H13O21N、C210H11O16N、C174H6O12N、C130H4O11和C94H3O15;在7 MPa压力下2 000 K、3 000 K、3 200 K、3 400 K、3 600 K和3 800 K温度对应的煤焦分子式分别为C268H30O15N2、C237H12O20N、C208H9O17N、C146H7O16N、C84H3O9N和C33H2O5;在10 MPa压力下2 000 K、3 000 K、3 200 K、3 400 K和3 600 K温度对应的煤焦分子式分别为C268H30O15N2、C202H7O27N、C169H4O22、C108O24和C49HO14。由此可见,随着温度的升高,煤焦分子中的C原子和H原子数量逐渐减少,而O原子数量基本上先增加后减少。在煤焦富氧燃烧过程中,首先,O2分子会吸附在煤焦表面的活性碳位点上,这是煤焦分子中O原子数量增加的主要原因;然后,与煤焦键结的氧会导致C—H或C—C键的断裂,其中C—H键断裂会形成OH自由基,C—C键断裂会导致苯环的开环反应并生成CO。10 MPa压力下煤焦中的O原子数量明显高于4 MPa和7 MPa压力下的O原子数量,而C原子和H原子数量却明显少于4 MPa和7 MPa压力下对应的原子数量。从微观分子层面的结果来看,增压提高了O2分子与煤焦分子的碰撞频率,从而促进了煤焦的氧化反应。另外,由于煤焦分子中活性碳位点的数量有限,增压促进了O2分子在煤焦表面的吸附,占据了更多的活性碳位点,抑制了CO2分子在煤焦表面的吸附,从而抑制了煤焦的气化反应。
(a)氧化反应

(a)4 MPa
3 结 论
(1)在4~10 MPa压力范围内,虽然增压降低了煤焦的气化反应速率,但是显著提高了煤焦的氧化反应速率,从而增加了煤焦的整体碳转化率。
(2)增压降低了O2和CO2分子的扩散系数,高压能促进煤焦氧化反应是由于煤焦周围较高的O2浓度;而CO2分子的扩散对气化反应的影响更为显著。
(3)4 MPa、7 MPa和10 MPa压力下煤焦氧化反应的活化能分别为151 kJ/mol、117 kJ/mol和81 kJ/mol,煤焦气化反应的活化能分别为174 kJ/mol、166 kJ/mol和172 kJ/mol。增压降低了煤焦氧化反应的活化能,但对煤焦气化反应活化能的影响并不明显。
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
材料与冶金学报(2022年2期)2022-08-10
北京航空航天大学学报(2022年7期)2022-08-06
安徽化工(2022年4期)2022-08-02
可再生能源(2022年6期)2022-06-22
黑龙江大学自然科学学报(2022年1期)2022-03-29
诗林(2019年6期)2019-11-14
中学物理·高中(2016年8期)2016-08-08
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
