
广东省侵染美香占2号的稻瘟病菌致病性及无毒基因变异分析
2022-04-14 01:41汪文娟苏菁陈深杨健源陈凯玲冯爱卿汪聪颖封金奇陈炳朱小源
中国农业科学 2022年7期
汪文娟,苏菁,陈深,杨健源,陈凯玲,冯爱卿,汪聪颖,封金奇,陈炳,朱小源
广东省侵染美香占2号的稻瘟病菌致病性及无毒基因变异分析
汪文娟,苏菁,陈深,杨健源,陈凯玲,冯爱卿,汪聪颖,封金奇,陈炳,朱小源
广东省农业科学院植物保护研究所/广东省植物保护新技术重点实验室,广州 510640
【】分析侵染广东稻区优质品种美香占2号稻瘟病菌()的致病性及无毒基因变异特征,为美香占2号在不同稻区的合理布局提供参考依据。利用9个抗稻瘟病单基因系,对稻瘟病菌单孢分离菌株进行致病性测定;并利用8个稻瘟病菌已克隆无毒基因的功能性分子标记对2013—2018年不同年份、不同稻区采集的稻瘟病菌单孢分离菌株的DNA进行PCR扩增,通过琼脂糖凝胶电泳检测分析,选取不同带型和不同年份代表性菌株的相应无毒基因全长或CDS区域的PCR片段进行测序。测序结果与相应无毒基因的参考序列进行碱基序列的比较分析;并利用相应的水稻抗稻瘟病单基因系,对无毒基因不同变异类型的稻瘟病菌菌株进行致病性测定。根据稻瘟病菌两种交配型MAT1-1和MAT1-2型分子标记,检测侵染美香占2号的稻瘟病菌可能存在的交配型。致病性分析结果显示,2013—2018年采集自美香占2号品种的52个稻瘟病菌菌株对其中的4个单基因系IRBL9-W()、IRBLzt-T()、NIL-e1()和IRBLkh-K3()均表现无毒;然而,侵染单基因系IRBLz-Fu()、IRBLkp-K60()和IRBLi-F5()的有毒菌株频率维持在57%以上,且侵染单基因系IRBLz-Fu()的有毒菌株频率在不同年份间呈现显著性差异,表明美香占2号种植区田间稻瘟病菌群体中存在较高频率的、和毒性菌株。根据8个已克隆无毒基因的功能性分子标记检测,在52个供试菌株中均可检测到无毒基因、和,但扩增不到、和,仅有4个菌株可检测到无毒基因,有3个菌株可检测到无毒基因。所测序7个菌株的编码区序列和包含启动子及编码区的序列与其各自无毒基因的序列完全一致;所测序7个菌株的包含启动子及编码区的序列中,有4个菌株的序列与无毒基因序列完全一致,有3个菌株在编码区存在CTTT连续4个碱基的插入。在来自广东不同稻区美香占2号的分离菌株群体中,共发现了4种不同的无毒基因扩增模式(扩增预期大小的条带、有转座子Pot3插入的条带、有转座子Mg-SINE插入的条带和无扩增条带),共呈现5种不同的单倍型(AvrPib-AP1-1、AvrPib-AP1-2、avrPib-AP2、avrPib-AP3和avrPib-AP2+avrPib-AP3),AvrPib-AP1-1和AvrPib-AP1-2是无毒型,而avrPib-AP2和avrPib-AP3则在基因的启动子区域分别存在Pot3或Mg-SINE的插入而呈现毒性型,在1个菌株中检测到avrPib-AP2+avrPib-AP3双单倍型。交配型分子标记分析结果表明,检测的52个稻瘟病菌菌株均为MAT1-2交配型,其中有2个菌株GD18-009和GD18-185采集于广东韶关稻区,同时可检测到MAT1-1交配型,具有双交配型。在广东美香占2号品种上分离的稻瘟病菌普遍含有无毒基因、、和,的变异类型丰富多样,该研究结果可为优质品种美香占2号的科学布局及其种植区域其他不同抗病基因型品种的合理选择提供参考。
美香占2号;稻瘟病菌;无毒基因型;交配型
0 引言
【研究意义】水稻()是世界上最重要的粮食作物之一,由子囊真菌稻瘟病菌()引起的稻瘟病是全球水稻生产上最具毁灭性的病害,显著降低了水稻产量和谷物质量[1-3]。尽管种植抗病品种是目前控制稻瘟病危害最经济、高效与环保的策略[4-5];但由抗瘟基因控制的抗病品种在推广种植3—5年后,常由于在寄主强大的选择压下,稻瘟病菌无毒基因的易变导致新的毒性小种产生及优势化而“丧失”抗瘟性,成为高度易感病的品种,引发稻瘟病的重新流行与暴发[6]。美香占2号是近年来华南食味品质优异、推广面积最大的优质稻品种[7]。随着该品种连续多年在生产上的大面积推广种植,其抗瘟性逐渐下降[8]。探明广东稻区侵染美香占2号的稻瘟病菌致病性、无毒基因类型及其变异情况,可为本稻区该品种的种植、抗病品种的轮换以及新抗病品种的选育提供科学依据。【前人研究进展】水稻和稻瘟病菌间的互作系统属于典型的“基因对基因”系统[9],只有在寄主植物中存在相应的抗病基因,而病原菌中又存在与之匹配的无毒基因时,寄主才表现出抗病性[10-11]。抗病基因和无毒基因的共同进化和相互作用促进了基因与基因间特异性“军备竞赛”模式的形成,从而导致抗病基因和无毒基因变异的多样性[12]。目前,水稻稻瘟病菌的无毒基因有9个被克隆[13-18]。有研究表明,水稻品种中抗病基因在田间是否有抗病功能,取决于其对应无毒基因在田间稻瘟病菌群体中的存在频率,这为基于无毒基因的分子诊断推断抗病基因的有效性提供了科学依据[19-21]。稻瘟病菌的无毒基因显示快速进化的特征,包括存在/缺失多态性[22-23],这使得稻瘟病菌能够克服水稻品种的抗性,导致病害流行。鉴定田间稻瘟病菌遗传变异的方法主要有rep-PCR[24-26]、扩增片段长度多态性[27]、无毒基因特异性标记[19,28]以及传统的鉴别品种[29]和具有单个主效抗病基因的单基因系鉴别品种表型鉴定等[30-33]。【本研究切入点】深入阐明源自优质水稻品种的稻瘟病菌小种变异特征,需要从病原菌致病型、无毒基因型变异及田间病原菌群体的交配型分析等方面开展研究。【拟解决的关键问题】利用9个水稻抗稻瘟病单基因系和感病对照品种丽江新团黑谷,对2013—2018年采集自美香占2号品种的52个稻瘟病菌菌株进行致病性分析,根据8个已克隆无毒基因(、、、、、及)功能性分子标记的检测,明确源自美香占2号品种的稻瘟病菌无毒基因或其等位基因的存在/缺失情况,并对扩增的无毒基因产物进行测序,比较分析测序菌株间无毒基因序列的变异特征;利用自然界中存在的稻瘟病菌两种交配型开发的分子标记,分析在广东不同稻作区采集自美香占2号品种稻瘟病菌的交配型。为广东稻区优质抗瘟品种的合理布局与稻瘟病的有效控制提供依据。
1 材料与方法
1.1 病原菌材料、繁殖及产孢
病原菌来源:2013—2018年从广东不同稻区的常规稻品种美香占2号上采集穗颈瘟标样,从中分离、纯化稻瘟病菌菌株。
田间采集的穗颈瘟标样,用0.5%的强氯精消毒2 min,随后用无菌水清洗,置于灭菌的培养皿中保湿培养约3 d,利用孢子振落法进行病原菌株的单孢分离。共分离获得稻瘟病菌单孢菌株52个(表1),所有菌株保存于广东省农业科学院植物保护研究所。

表1 采自广东的52 个稻瘟病菌菌株信息
菌株活化及产孢:将保存的菌株转移至酵母固体培养基,25℃左右的生化培养箱中活化培养7 d以上;然后将菌丝体转接到玉米培养基上繁殖,25℃左右培养13 d。用消毒过的无菌水洗去玉米粒表面的菌丝,将玉米粒置于消毒的搪瓷盘中(25 cm×19 cm×2 cm),上面覆盖一层湿纱布,然后在日光灯下光照培养3—4 d,进行产孢。
1.2 致病性测定
植物材料:9个抗稻瘟病单基因系分别为IRBL9-W()、IRBLzt-T()、NIL-e1()、IRBLkh-K3()、IRBL1-CL()、IRBLta2-Re(Pita)、IRBLz-Fu()、IRBLkp-K60()、IRBLi-F5(),其中NIL-e1是广东省农业科学院植物保护研究所以丽江新团黑谷为背景,育成的含0的抗稻瘟病近等基因系[34],其余单基因系由国际水稻研究所选育。感病对照为丽江新团黑谷(LTH)。
材料种植:将水稻种子催芽后穴播于30 cm×20 cm×5 cm规格的搪瓷盆里,每盆播12个材料,每个材料播种量约为10粒种子,设置3次重复。稻苗采取旱育栽培,长至1叶1心期用硫酸铵施肥,每盆施肥量为0.5 g,接种前共需施肥3次。待稻苗长至3.5—4叶龄时,进行人工喷雾接种。
接种及调查:用无菌水洗下玉米粒上的孢子,用两层塑料细纱网隔去玉米残渣,配成适量浓度的孢子液,一般将孢子液浓度调至1×105个/mL,进行人工喷雾接种。接种后置于遮光密闭的培养箱中暗培养,在25℃及相对湿度达95%以上的条件下保湿24 h,之后转至玻璃温室,在25—28℃下保湿培养至稻苗发病,接种7 d左右调查稻苗的整体发病情况。病级调查方法按照国际水稻研究所稻瘟病圃苗瘟分级标准进行:0—3级为抗病(R);4—9级为感病(S)。
1.3 稻瘟病菌DNA的提取
将分离纯化的稻瘟病菌单孢菌株在CM(完全培养基)固体培养基上活化培养,挑取适量菌丝块接到CM液体培养基中,于28℃摇床,160 r/min振荡培养3—5 d,收集菌丝体;用Omega公司的真菌DNA抽提试剂盒提取菌丝体的基因组DNA。
1.4 无毒基因分子标记和交配型检测
引物设计:8个已克隆稻瘟病菌的无毒基因(、、、、、、和)中,和分子标记设计分别根据NCBI GenBank中(GenBank 登录号:KM887844.1)和(Genbank登录号:EU837058.1)序列,利用软件 Primer Premier 5.0,设计覆盖和启动子和开放阅读框的区域。其他6个无毒基因的特异引物参照Selisana等[19]开发的标记,交配型分析分子标记参照Olukayode等[20]。所有引物均在生工生物工程(上海)股份有限公司(广州生工)合成,引物序列详见表2。
无毒基因分子标记检测:PCR反应总体积为20 µl,其中包括稻瘟病菌基因组DNA(20—30 ng·μl-1)1 μl;10×PCR Mixture 10 μl,正、反向引物(10 μmol·L-1)各0.5 μl,ddH2O 8 μl。PCR扩增程序:94℃预变性3 min,94℃变性30 s,58—60℃(根据不同引物而定)退火30 s,72℃延伸1 min/kb,共设置35次循环,最后72℃延伸5 min。PCR产物于1%—1.5%的琼脂糖凝胶中电泳检测。
1.5 部分无毒基因序列测序分析
利用高保真酶KOD-Plus(东洋纺,日本)进行PCR扩增,并对PCR产物进行目的片段的纯化,将纯化的PCR产物送到生工生物工程(上海)股份有限公司(广州生工)进行测序。采用Lasergene7.0软件中的SeqMan (http://www.dnastar.com/)进行测序序列的多序列比对拼接;并采用DNAStar MegAlign Clustal V软件对有差异的核苷酸序列进行比较分析。
2 结果
2.1 分离自美香占2号菌株对部分水稻抗稻瘟病单基因系的致病性
利用9个水稻抗稻瘟病单基因系和感病对照品种丽江新团黑谷,对2013—2018年采集自美香占2号品种的52个稻瘟病菌菌株进行了致病性分析,其中由于2014年采集菌株数较少,未进行结果统计。结果如表3所示,所测定的菌株对其中的4个单基因系IRBL9-W()、IRBLzt-T()、NIL-e1()和IRBLkh-K3()均表现无毒。2013—2018年间,侵染单基因系IRBL1-CL()的有毒菌株频率在不同年份间呈现极显著性差异;侵染单基因系IRBLta2-Re(Pita)和IRBLz-Fu()的有毒菌株频率不同年份间均呈现显著性差异;侵染单基因系IRBLkp-K60()IRBLi-F5()的有毒菌株频率在不同年份间差异不显著。上述结果表明,在美香占2号品种种植区,可能由于田间稻瘟病菌群体中存在较高频率的和的毒性菌株,且在2013—2018年间,和avrPita的毒性菌株频率的不断累积,导致美香占2号品种的抗性有效性逐渐减弱。

表2 本研究中用于检测无毒基因和交配型的引物

表3 2013—2018年采集的稻瘟病菌群体中对抗病单基因系有毒菌株频率
星号表示参数值差异显著 Theasterisk indicated the values of the parameter differed significantly。 *:<0.05,df=4;**:0.0001<<0.05,df=4;***:<0.0001,df=4
2.2 美香占2号分离菌株含有的无毒基因型
为了确定试验菌株中8个已克隆无毒基因的单倍型,利用Selisana等[19]开发的6个无毒基因分子标记及本研究室设计开发的[21]和分子标记(表2),通过PCR扩增,对52个供试菌株进行无毒基因单倍型分析。部分菌株无毒基因的PCR扩增结果如图1所示。在52个供试菌株中均能检测到无毒基因和的基因片段;相反地,在任何菌株中都扩增不到、和。其中只有4个菌株GD13-452、GD14-295、GD18-009和GD18-185可检测到无毒基因的基因片段;仅有3个菌株GD16-034、GD18-009和GD 18-185可检测到无毒基因的基因片段。启动子和CDS序列的扩增和测序结果发现,无毒基因在所检测的52个菌株中,共发现了4种不同的扩增模式,分别为扩增预期大小的条带(AP1)、有转座子Pot3插入的条带(AP2)、有转座子Mg-SINE插入的条带(AP3)和无扩增条带(AP4)(图2-a、2-b)。AP1表示与扩增参考序列的无毒菌株CHL42(Genbank登录号:KM887844.1)大小一致的扩增子,AP2和AP3分别代表大约1.0和2.5 kb的扩增子,比无毒分离株的预期大小要大(图2-b),AP4表示扩增无带,完全缺失。在52个广东稻区分离菌株中,分别有5、12、33、1和1个菌株可检测到AP1、AP2、AP3、AP2+AP3和AP4型基因型,检测频率分别为9.62%、23.08%、63.46%、1.92%和1.92%。
对GD13-452、GD14-295、GD15-196、GD15-205、GD16-034、GD18-009和GD18-185共7个菌株进行、和相应无毒基因的全长或CDS区域PCR产物测序。结果表明,所测序7个菌株的编码区序列和包含启动子及编码区的序列,与其各自无毒基因的序列完全一致(Genbank登录号分别为KM004023.1和EU837058.1);所测序的7个菌株的包含启动子及编码区的序列中,有4个菌株的序列与无毒基因序列完全一致(Genbank登录号:AB498875.1),有3个菌株(GD14-295、GD15-196、GD15-205)在编码区存在CTTT连续4个碱基的插入(结果未显示)。

M:Marker,DL2000或DL500;1—16:部分试验菌株的DNA Some DNA samples of test strains
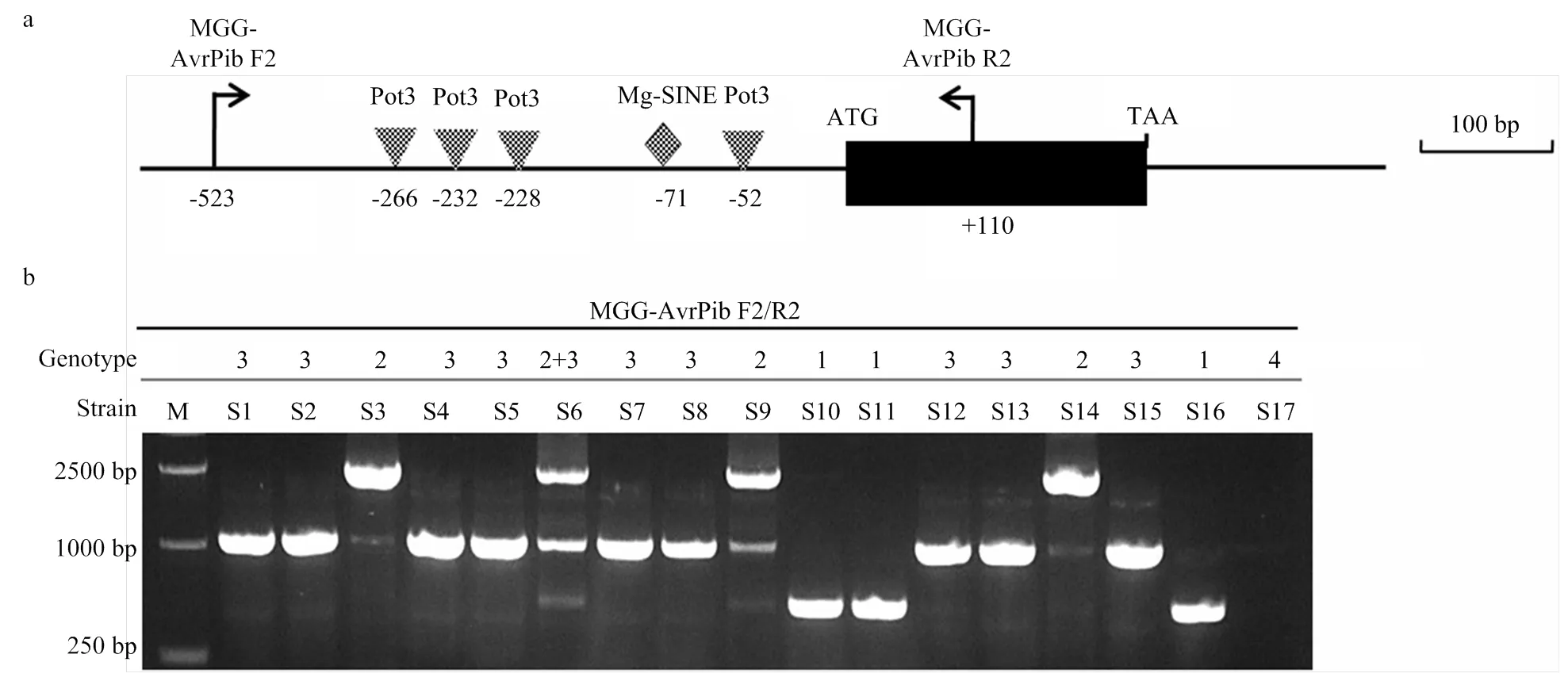
a:引物MGG-AvrPibF2/R2检测转座子Pot3和 Mg-SINE在AvrPib(KM887844.1)中的插入The positions of the Pot3 and Mg-SINE transposon detected by the primer pair MGG-AvrPibF2/R2 within the AvrPib sequence (KM887844.1);b:17 个测序稻瘟病菌菌株的基因型,1—3(1条可见带型)、4(缺失)Genotyping of 17 sequenced M. oryzae strains. 1-3 (one fragment of variable size), 4 (absent)
2.3 分离自美香占2号稻瘟病菌中无毒基因AvrPib等位基因单倍型的序列多样性
为了进一步分析分离自美香占2号品种的稻瘟病菌等位基因的遗传多样性及其变异情况,选择含有不同单倍型的15个代表菌株,对其扩增产物进行序列分析。结果表明,4个测序的AP1型菌株鉴定出了两种源自AP1的不同单倍型,分别称为和,与的参考序列相比,型仅在启动子区域的两个位置存在序列差异,即是-216 bp处5′-C-3′的插入和-124 bp处5′-AAC-3′的插入,其中有一个菌株GD13-394属于AvrPib-AP1-2型,除了上述两个插入,在-269 bp处还存在5′-TAAC-3′的插入(图3-a)。源自AP2的单倍型,被称为,所测序的5个菌株中,分别在-266、-232、-228和-52 bp处存在Pot3转座子的插入,该Pot3转座子的序列与NCBI公开的序列(Genbank登录号:U60989.1)具有100%的核苷酸序列同源性,其中一个菌株GD16-196在-269 bp处还检测到5′-TAAC-3′的插入。与不同,来自AP3的单倍型,被称为,在-71 bp的位置存在Mg-SINE转座子的插入,该Mg-SINE转座子的序列与NCBI公开的序列(Genbank登录号:U35313.1)具有100%的核苷酸序列同源性;除了Mg-SINE转座子插入之外,在启动子区域的其他两个位置也含有与单倍型相同的序列突变(图3-a)。Pot3转座子的侧翼为靶位点重复(TSD)的两个碱基对序列(5′-TA-3′),在两个菌株中-269 bp检测到的5′-TAAC-3′插入序列,可以假定是从Pot3转座子的插入位置切除了Pot3的插入序列引起的(图3-a)。上述结果表明,转座子的插入或切除可能在田间分离菌株中呈动态的变化。
2.4 分离自美香占2号稻瘟病菌不同AvrPib等位基因单倍型的致病性及出现频率
为了明确不同等位基因单倍型菌株对单基因系IRBLb-B()的无毒/有毒性反应,选取上述15个测序代表菌株,进行单基因系IRBLb-B()的苗期致病性测定。结果如图3-b、表4所示,AvrPib-AP1分离菌株显示出对IRBLb-B的无毒性反应,表明它们都能够触发介导的免疫反应。相反地,AvrPib-AP2和AvrPib-AP3分离菌株对IRBLb-B具有毒性反应,表明它们没有触发介导的免疫反应。因此,将这两种单倍型重命名为avrPib-AP2和avrPib-AP3。进一步分析了15个菌株中这3种单倍型的出现频率,发现AvrPib-AP1-1、AvrPib-AP1-2、avrPib-AP2、avrPib-AP3和avrPib-AP2+avrPib-AP3单倍型菌株的出现频率分别为20.00%、6.67%、33.33%、33.33%和6.67%(表4)。
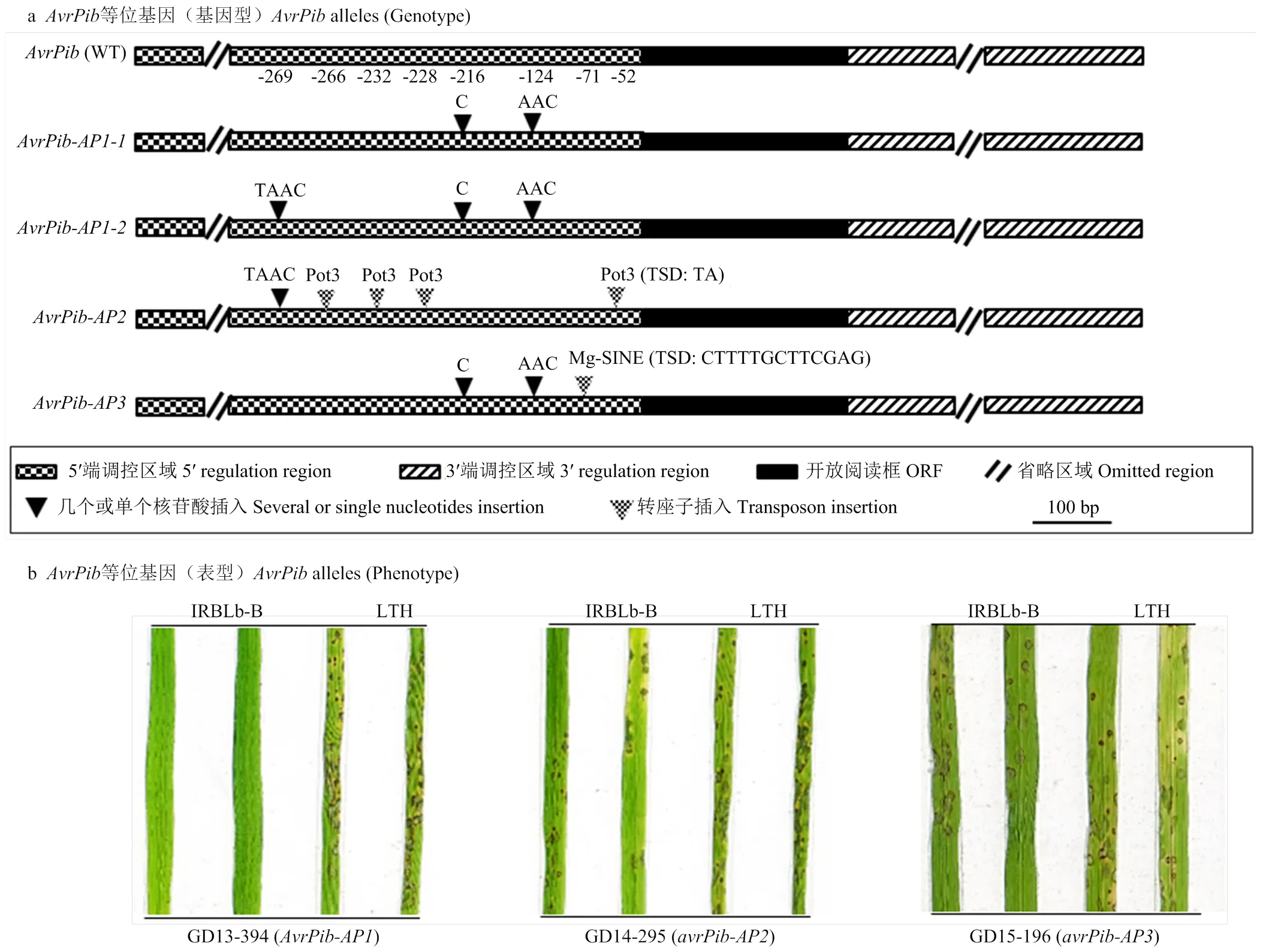
a:AvrPib等位基因变异特征Characterization of allelic variations at AvrPib。WT:野生型 Wild type (AvrPib-KM887844.1);AP1-1:TAACGT插入 TAACGT insertion;AP1-2:TAAC 插入 TAAC insertion;AP2:Pot3插入 Pot3 insertion;AP3:Mg-SINE 插入 Mg-SINE insertion。b:具有不同AvrPib等位基因变异型菌株的表型The phenotype of strains with different AvrPib allelic variations

表4 15个代表菌株的AvrPib单倍型特征
a基因位置是基于参考序列的位置The positions were based on the reference sequence of(GenBank accession number: KM887844.1)
2.5 分离自美香占2号稻瘟病菌群体的交配型分析
为了分析不同年份在广东省不同稻作区采集的美香占2号侵染菌株的交配型,对52个菌株利用自然界中存在的两种稻瘟病菌交配型MAT1-1和MAT1-2开发的分子标记进行分析。结果表明,选择的52个稻瘟病菌株均为MAT1-2交配型,其中有两个菌株GD18-009和GD18-185同时可检测到MAT1-1交配型,具有双交配型,这两个菌株在广东省韶关稻区采集(部分菌株的检测结果如图4所示)。

M:Marker,DL2000;1—15:部分试验菌株DNA some DNA samples of test strains
3 讨论
3.1 无毒基因监测与品种布局
基于抗病单基因系对稻瘟病菌无毒基因进行监测,结合已克隆无毒基因的分子标记分析病原菌的无毒基因型,可有效地用于稻瘟病菌无毒基因的精准分析及对水稻品种有效抗病基因的科学推断[32-33]。目前,水稻抗稻瘟病近等基因系和单基因系已成为稻瘟病菌致病性及无毒基因分析的主要鉴别品种[30,35]。本研究所选用的一套由9个抗病单基因系构成的稻瘟病菌单基因系鉴别品种,其对我国华南籼稻区稻瘟病菌的致病性有较强的鉴别力[36]。本研究的单基因系监测结果表明,所检测的52个稻瘟病菌菌株对其中的4个单基因系IRBL9-W()、IRBLzt-T()、NIL-e1()、IRBLkh-K3()均无毒。基于无毒基因分子标记的检测表明,在52个稻瘟病菌菌株中均可检测到、和-型无毒基因,上述抗稻瘟病单基因系的监测结果与无毒基因型分析的结果相一致。据报道,具有的6个不同等位基因(A、B、C、D、E和F型),主要突变类型是基因编码区单碱基的突变引起氨基酸的变异,共存在5个氨基酸突变位点(46、47、48、67和78)[13],-型可被抗病基因和所识别,-型可被抗病基因和识别,-型可被抗病基因所识别[37]。在2013—2018年间分离的52个稻瘟病菌菌株中,侵染单基因系IRBLkp-K60()的有毒菌株频率66.67%—88.89%,且不同年份间差异不显著;然而,在所有的菌株中都可检测到-基因片段,推断所检测的目的基因片段大多属于型或型,该推断需要进一步对-扩增片段进行测序验证。
本研究的单基因系监测结果显示,侵染单基因系IRBLz-Fu()和IRBLi-F5()的有毒菌株频率分别维持在57%和72%以上,表明美香占2号种植区田间稻瘟病菌群体中对抗病基因和具有毒性的菌株存在频率较高。在2013—2018年间,侵染单基因系IRBL1-CL()和IRBLta2-Re(Pita)的有毒菌株频率较低,在34.0%以下;基于无毒基因的分子标记检测,在52个稻瘟病菌菌株中也检测不到无毒基因。由于无毒基因、AvrPita和尚未被克隆,因此未进行相应无毒基因型的检测。本研究利用抗瘟基因和基因簇分子标记、和基因特异性分子标记检测等对美香占2号品种含有的抗病基因进行检测,结果均未检测到抗病基因、及、抗病基因簇的等位基因(结果未显示),推测美香占2号品种可能含有其他未被克隆的抗病基因。
综上所述,根据侵染美香占2号的稻瘟病菌菌株对水稻抗瘟单基因系和无毒基因分子标记的检测结果,在美香占2号感病区域应避免种植含抗病基因、和的水稻品种,可轮换种植含、、或等抗病基因的水稻品种,该结论与本研究团队前期对广东部分稻区侵染美香占2号稻瘟病菌致病性分析的结果相一致[8]。
3.2 稻瘟病菌无毒基因变异机制
为了抑制或逃避寄主植物的识别,稻瘟病菌常通过无毒基因的突变包括点突变、在基因或启动子区域插入转座子以及部分或全部删除无毒基因从而导致无毒基因的功能丧失[15,38]。这些无毒基因的突变机制与抗病基因-无毒基因间协同进化的历史、无毒基因的适应度及其在病原菌基因组中的位置相关。据报道,无毒基因与等位基因间呈现竞争性的阶梯型共进化模式[37]。Li 等[38]分析中国云南省稻瘟病菌群体发现,自然界正向选择压下共鉴定到5种新的变异单倍型。无毒基因的等位基因呈高度多样化,包括编码区的部分缺失或完全缺失、点突变、移码突变,启动子区的转座子插入等[24]。
关于无毒基因的突变分析,Zhang等[16]报道,在中国分离株中发现了基因片段缺失、点突变、TE插入和完全缺失4种变异,所有这些变异均导致了中国5个不同稻区稻瘟病菌群体对抗病基因的无毒性功能丧失。Olukayode等[20]分析了菲律宾田间稻瘟病菌群体的基因序列变异,发现中的Pot3转座子插入导致稻瘟病菌对含的水稻品种具有毒性,并在3个不同的基因组位置检测到 Pot3插入,导致3个不同的单倍型。孟峰等[39]在黑龙江省稻瘟病菌中检测出4种带型(无带、高带、中高带与低带)和5种基因型,未检测到双带型。Xiao等[40]在27个代表菌株组成的稻瘟病菌群体中,基于PCR扩增子的序列鉴定了9种AvrPib不同的单倍型,即AvrPib-H1至AvrPib-H9型,其中AvrPib-H1与AvrPib(Genbank登录号:KM887844)相同,AvrPib-H2、AvrPib-H4和Avr-Pib-H9与Zhang等[16]报道的相同。然而,AvrPib-H3在+97位(Genbank登录号:MN630588)包含从A到T的单核苷酸取代,而AvrPib-H5至AvrPib-H8分别在-56、-274、-65、-125 bp处包含一个Pot3插入。Wang等[21]研究发现,在广东韶关新银占单一品种种植区采集的稻瘟病菌群体中,共发现了无毒基因4种不同的单倍型(AvrPib-AP1 -1、AvrPib-AP1-2、avrPib-AP2和avrPib-AP3),分别在上游的-266和-87 bp处检测到转座子Pot3或Mg-SINE的插入。本研究分别在上游的-266、-232、-228和-52 bp检测到转座子Pot3插入,在-71 bp处检测到转座子Mg-SINE插入;检测到的转座子插入位置具有更丰富的多样性,这可能与稻瘟病菌虽都采集自单一品种美香占2号,但却从广东省不同的稻区采集相关。本研究中,在广东省不同稻区美香占2号分离菌株群体中共发现了无毒基因4种不同的扩增模式:扩增预期大小的条带、有转座子Pot3插入的条带、有转座子Mg-SINE插入的条带和无扩增条带,共呈现5种不同的单倍型(AvrPib-AP1-1、AvrPib-AP1-2、avrPib-AP2、avrPib-AP3和avrPib-AP2+avrPib-AP3),AvrPib-AP1-1和AvrPib-AP1-2是无毒型的,而avrPib-AP2和avrPib-AP3则在基因的启动子区域分别存在Pot3或Mg-SINE的插入而呈现毒性型,在一个菌株中检测到avrPib-AP2+avrPib-AP3双单倍型。本研究与Wang等[21]的研究中均在启动子区域的两个相同的位置检测到单个或者连续几个碱基的插入(-216 bp处5′-C-3′的插入和-124 bp处5′-AAC-3′的插入),推断这两个位点的突变可能是个古老存在的突变位点,在抗病品种引入之前该两个位点的突变已经存在,由于该位点的突变不引起稻瘟病菌无毒基因功能的改变,从而在进化过程中得以保留。
4 结论
美香占2号种植区田间稻瘟病菌群体中普遍含有无毒基因、、和,而不含无毒基因、和在美香占2号感病区域可轮换种植含、、或等抗病基因的水稻品种。在美香占2号分离菌株群体中,无毒基因的变异类型具有丰富的多样性,共发现4种不同的扩增模式,显示5种不同的单倍型。启动子区域分别存在Pot3或Mg-SINE的插入是导致无毒性功能丧失的主要机制,且转座子的插入或切除可能在本试验菌株中呈动态变化。侵染美香占2号的稻瘟病菌在田间主要以无性繁殖为主,绝大部分菌株只检测到MAT1-2型,交配型较单一,在广东韶关稻区检测到MAT1-1和MAT1-2双交配型的菌株,表明广东韶关稻区田间稻瘟病菌交配型较丰富,该稻区稻瘟病菌群体可能存在着丰富的遗传多样性。
[1] KHUSH G S, Jena K K. Current status and future prospects for research on blast resistance in rice (L.)//Wang G L, VALENT B. Advances in genetics, genomics and control of rice blast disease. New York: Springer, 2009: 1-10.
[2] Zhang H f, Zheng X b, Zhang Z g. Thespecies complex and plant pathogenesis. Molecular Plant Pathology,2016, 17(6): 796-804.
[3] Savary S, Willocquet L, Pethybridge S J, Esker P, McRoberts N, Nelson A. The global burden of pathogens and pests on major food crops. Nature Ecology and Evolution, 2019, 3(3): 430-439.
[4] Ashkani S, Rafii M Y, Shabanimofrad M, Miah G, Sahebi M, Azizi P, Tanweer F A, Akhtar M S, Nasehi A. Molecular breeding strategy and challenges towards improvement of blast disease resistance in rice crop. Frontiers in Plant Science, 2015, 6: 886.
[5] TANWEER F A, RAFII M Y, SIJAM K, RAHIM H A, AHMED F, LATIF M A. Current advance methods for the identification of blast resistance genes in rice. Comptes Rendus Biologies, 2015, 338(5): 321-334.
[6] Kirzinger M W, Stavrinides J. Host specificity determinants as a genetic continuum. Trends in Microbiology, 2012, 20(2): 88-93.
[7] 赵雷, 周少川, 王重荣, 李宏, 黄道强, 王志东, 周德贵, 陈宜波, 吴玉坤. 绿色广适性优质稻品种的系谱分析及育种应用研究. 生命科学, 2018, 30(10): 1100-1107.
ZHAO L, ZHOU S C, WANG C R, LI H, HUANG D Q, WANG Z D, ZHOU D G, CHEN Y B, WU Y K. Pedigree analysis and breeding and application of green widely adaptive good quality rice varieties. Chinese Bulletin of Life Sciences, 2018, 30(10): 1100-1107. (in Chinese)
[8] 汪文娟, 苏菁, 杨健源, 陈深, 鲁国东, 朱小源. 侵染优质稻美香占2号的稻瘟病菌生理小种鉴定及无毒基因分析. 植物保护学报, 2020, 47(3): 572-582.
WANG W J, SU J, YANG J Y, CHEN S, LU G D, ZHU X Y. Identification of physiological race and analysis avirulent genes for isolates of rice blast infecting from rice variety of Meixiangzhan 2. Journal of plant protection, 2020, 47(3): 572-582. (in Chinese)
[9] FLOR H H. Current status of the gene-for-gene concept. Annual Review of Phytopathology, 1971, 9: 275-296.
[10] Orbach M j, Farrall L, Sweigard J a, Chumley F g, Valent B. A telomeric avirulence gene determines efficacy for the rice blast resistance gene. The Plant Cell, 2000, 12(11): 2019-2032.
[11] Valent B, Khang C h. Recent advances in rice blast effector research. Current Opinion in Plant Biology, 2010, 13(4): 434-441.
[12] Dodds P n, Lawrence G j, Catanzariti A m, Teh T, Wang C a, Ayliffe M a, Kobe B, Ellis J g. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proceedings of National Academy of Sciences of the United States of America, 2006, 103(23): 8888-8893.
[13] Yoshida K, Saitoh H, Fujisawa S, Kanzaki H, Matsumura H, Yoshida K, Tosa Y, Chuma I, Takano Y, Win J, Kamoun S, Terauchi R. Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen. The Plant Cell, 2009, 21(5): 1573-1591.
[14] Kanzaki H, Yoshida K, Saitoh H, Fujisaki K, Hirabuchi A, Alaux L, Fournier E, Tharreau D, Terauchi R. Arms race co-evolution ofand ricegenes driven by their physical interactions. The Plant Journal, 2012, 72(6): 894-907.
[15] Wu J, Kou Y j, Bao J d, Li Y, Tang M z, Zhu X l, Ponaya A, Xiao G, Li J b, Li C y,. Comparative genomics identifies theavirulence effectorthat triggers-mediated blast resistance in rice. New Phytologist, 2015, 206(4): 1463-1475.
[16] Zhang S l, Wang L, Wu W h, He L y, Yang X f, Pan Q h. Function and evolution ofavirulence generesponding to the rice blast resistance gene. Scientific Reports, 2015, 5: 11642.
[17] Ray S, Singh P k, Gupta D k, Mahato A k, Sarkar C, Rathour R, Singh N k, Sharma T r. Analysis ofgenome reveals a fungal effector, which is able to induce resistance response in transgenic rice line containing resistance gene,. Frontiers in Plant Science, 2016, 7: 1140.
[18] Wang B h, Ebbole D j, Wang Z h. The arms race betweenand rice: diversity and interaction ofandgenes. Journal of Integrative Agriculture, 2017, 16(12): 2746-2760.
[19] Selisana S m, Yanoria M j, Quime B, Chaipanya C, Lu G, Opulencia R, Wang G l, Mitchell T, Correll J, Talbot N j, Leung H, Zhou B. Avirulence () gene-based diagnosis complements existing pathogen surveillance tools for effective deployment of resistance () genes against rice blast disease. Phytopathology, 2017, 107(6): 711-720.
[20] Olukayode T, Quime B, Shen Y c, Yanoria M j, Zhang S b, Yang J y, Zhu X y, Shen W c, vON Tiedemann A, Zhou B. Dynamic insertion of Pot3 inprevailing in a field rice blast population in the Philippines lead to the high virulence frequency against the resistance genein rice. Phytopathology, 2019, 109(5): 870-877.
[21] Wang W J, Su J, Chen K L, Yang J Y, Chen S, Wang C Y, Feng A Q, Wang Z H, Wei X Y, Zhu X Y, Lu G D, Zhou B. Dynamics of the rice blast fungal population in the field after deployment of an improved rice variety containing known resistance genes. Plant Disease, 2021, 105(4): 919-928.
[22] Latorre S M, Reyes-Avila C S, Malmgren A, Win J, Kamoun S, Burbano H A. Differential loss of effector genes in three recently expanded pandemic clonal lineages of the rice blast fungus. BMC Biology, 2020, 18(1): 88.
[23] Yoshida K, Saunders D G, Mitsuoka C, Natsume S, Kosugi S, Saitoh H, Inoue Y, Chuma I, Tosa Y, Cano L M, Kamoun S, Terauchi R. Host specialization of the blast fungusis associated with dynamic gain and loss of genes linked to transposable elements. BMC Genomics, 2016, 17: 370.
[24] Xing J, Jia Y, Correll J C, Lee F N, Cartwright R, Cao M, Yuan L. Analysis of genetic and molecular identity among field isolates of the rice blast fungus with an international differential system, Rep-PCR, and DNA sequencing. Plant Disease, 2013, 97(4): 491-495.
[25] Chen Q H, Wang Y C, Zheng X B. Genetic diversity ofin China as revealed by DNA fingerprint haplotypes and pathotypes. Journal of Phytopathology, 2006, 154(6): 361-369.
[26] George M L, Nelson R J, Zeigler R S, Leung H. Rapid population analysis ofby using rep-PCR and endogenous repetitive DNA sequences. Phytopathology, 1998, 88(3): 223-229.
[27] Thuan N T N, Bigirimana J, Roumen E D, VAN DER Straeten D, Höfte M. Molecular and pathotype analysis of the rice blast fungus in North Vietnam.European Journal of Plant Pathology, 2006, 114(4): 381-396.
[28] Imam J, Alam S, Mandal N P, Shukla P, Sharma T r, Variar M. Molecular identification and virulence analysis ofgenes in rice blast pathogen,from Eastern India. Euphytica, 2015, 206: 21-31.
[29] Le M T, Arie T, Teraoka T. Population dynamics and pathogenic races of rice blast fungus,in the Mekong Delta in Vietnam. Journal of General Plant Pathology, 2010, 76: 177-182.
[30] Tsunematsu H, Yanoria M J T, Ebron L A, Hayashi N, Ando I, Kato H, Imbe T, Khush G S. Development of monogenic lines of rice for rice blast resistance. Breeding Science, 2000, 50(3): 229-234.
[31] Wang J C, Correll J C, Jia Y. Characterization of rice blast resistance genes in rice germplasm with monogenic lines and pathogenicity assays. Crop Protection, 2015, 72: 132-138.
[32] Wang J C, Jia Y, Wen J W, Liu W P, Liu X M, Li L, Jiang Z Y, Zhang J H, Guo X L, Ren J P. Identification of rice blast resistance genes using international monogenic differentials. Crop Protection, 2013, 45: 109-116.
[33] Zhang Y L, Wang J Y, Yao Y X, Jin X H, Correll J, Wang L, Pan Q H. Dynamics of race structures of the rice blast pathogen population in Heilongjiang province, China from 2006 through 2015. Plant Disease, 2019, 103(11): 2759-2763.
[34] ZHU X Y, CHEN S, YANG J Y, ZHOU S C, ZENG L X, HAN J L, SU J, WANG L, PAN Q H. The identification of(t), a new member of the rice blast resistancemultigene family. Theoretical and Applied Genetics, 2012, 124(7): 1295-1304.
[35] 马军韬, 张国民, 张丽艳, 邓凌韦, 王永力, 王英. 黑龙江省水稻种质抗瘟性及稻瘟病菌致病性分析. 植物保护学报, 2017, 44(2): 209-216.
MA J T, ZHANG G M, ZHANG L Y, DENG L W, WANG Y L, WANG Y. Analysis of blast-resistance of rice germplasm and pathogenicity of rice blast fungusin Heilongjiang Province. Journal of Plant Protection, 2017, 44(2): 209-216. (in Chinese)
[36] 朱小源, 杨祁云, 杨健源, 雷财林, 王久林, 凌忠专. 抗稻瘟病单基因系对籼稻稻瘟病菌小种鉴别力分析. 植物病理学报, 2004, 34(4): 361-368.
ZHU X Y, YANG Q Y, YANG J Y, LEI C L, WANG J L, LING Z Z. Differentiation ability of monogenic lines toinrice. Acta Phytopathologica Sinica, 2004, 34(4): 361-368. (in Chinese)
[37] WU W H, WANG L, ZHANG S, LIANG Y Q, ZHENG X L, YI K X, HE C P. Assessment of sensitivity and virulence fitness costs of thealleles fromto isoprothiolane. Genetics and Molecular Research, 2014, 13(4): 9701-9709.
[38] Li J, Wang Q, Li C, Bi Y, Fu X, Wang R. Novel haplotypes and networks ofalleles in. BMC Plant Biology, 2019, 19: 204.
[39] 孟峰, 张亚玲, 靳学慧, 张晓玉, 姜军. 黑龙江省稻瘟病菌无毒基因、和的检测与分析. 中国农业科学, 2019, 52(23): 4262-4273.
MENG F, ZHANG Y L, JIN X H, ZHANG X Y, JIANG J. Detection and analysis ofavirulence genes,andin Heilongjiang Province. Scientia Agricultura Sinica, 2019, 52(23): 4262-4273. (in Chinese)
[40] Xiao G, Yang J y, Zhu X y, Wu J, Zhou B. Prevalence of ineffective haplotypes at the rice blast resistance () gene loci in Chinese elite hybrid rice varieties revealed by sequence-based molecular diagnosis. Rice, 2020, 13(1): 6.
pathogenicity and Avirulence Genes Variation offrom a Rice Variety Meixiangzhan 2 in Guangdong Province
WANG WenJuan, SU Jing, Chen Shen, Yang JianYuan, CHEN KaiLing, FENG AiQing, Wang CongYing, Feng JinQi, Chen Bing, ZHU XiaoYuan
Plant Protection Research Institute, Guangdong Academy of Agricultural Sciences/Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangzhou 510640
【】The objective of this study is to analyze the pathogenicity and variation patterns of avirulence genes (genes) genotype of, which was collected from a high-quality rice variety Meixiangzhan 2 widely cultivated in Guangdong, and to provide a reference for the rational layout of Meixiangzhan 2 in different rice ecological areas.【】The pathogenicity of single-spore strains was determined using 9monogenic differentials. The DNA of the single-sporestrains collected from Meixiangzhan 2 in different rice areas and in different years from 2013 to 2018 was subjected to PCR amplification using the functional markers of 8genes. The PCR products ofgenes with promotor sequences or CDS regions were analyzed by agarose gel electrophoresis and the PCR products of representative strains were sequenced, and their sequences were compared to correspondinggenes, respectively. The correspondingmonogenic differentials were used to determine the pathogenicity ofstrains with different mutation types of thegenes. According to two mating types MAT1-1 and MAT1-2 molecular markers of, the possible mating type ofstrains isolated from Meixiangzhan 2 was detected.【】The pathogenicity analysis with a set of monogenic differentials showed that 52 rice blast strains collected from Meixiangzhan 2 from 2013 to 2018 were avirulent to IRBL9-W (), IRBLzt-T (), NIL-e1 () and IRBLkh-K3 (). The tested strains showed high frequencies (>57%) of virulence to IRBLz-Fu (), IRBLkp-K60 () and IRBLi-F5 (), and the frequency of virulence to IRBLz-Fu () showed significant differences in different years, suggesting that thepopulation in the field of Meixiangzhan 2 planted area had a higher frequency of virulent strains of,and.,andfragments were almost present in all tested strains, but none of,orwas amplified in any strain. Only 4 strains could amplify the fragment of, and only 3 strains could amplify the fragment of. Amplicon sequencing of the 7 strains revealed that the sequences ofandwere identical to those of the correspondinggenes. Of the 7 sequenced strains in, 4 strains were almost identical to each other, and 3 strains had a ‘CTTT’ in the coding region of.Four different amplification types of(expected size bands, bands with transposon Pot3 insertion, bands with transposon Mg-SINE insertion, and non-amplified bands) were detected by electrophoresis analysis and 5 different haplotypes (AvrPib-AP1-1, AvrPib-AP1-2, avrPib-AP2, avrPib-AP3 and avrPib-AP2+avrPib-AP3) were detected by sequencing PCR products. The genotypes AvrPib-AP1-1 and AvrPib-AP1-2 were avirulent, while avrPib-AP2 and avrPib-AP3 showing insertions of transposon Pot3 or Mg-SINE with different insertion sites were virulent. A double haplotype of avrPib-AP2+avrPib-AP3 was detected in one strain. The result of mating type analysis with molecular markers showed that all of 52 tested strains belonged to the mating type of MAT1-2, two strains (GD18-009 and GD18-185) from Shaoguan, Guangdong were also detected with MAT1-1 mating type, indicating that these two strains had dual mating types.【】Among the strains from the high-quality rice variety of Meixiangzhan 2 in Guangdong, thegenes of,,anddistribute widely. The variation types ofare abundant. The results of this study will provide useful information for the rational layout of Meixiangzhan 2 and the deployment of other rotation varieties with different genotypes of rice blast resistance.
Meixiangzhan 2;; genotype of avirulence gene; mating type

10.3864/j.issn.0578-1752.2022.07.007
2021-11-01;
2021-12-20
国家重点研发计划(2016YFD0300707)、广东省自然科学基金(2020A1515011213)、广东省农业科学院“优秀博士”人才引进项目(R2021YJ-YB3020)、广东省农业科学院学科团队建设项目(202116TD)、国家现代农业产业技术体系建设专项(CARS-01-35)、广东省现代农业产业技术体系(2021KJ105)、广州市科技计划(202002030001)
汪文娟,E-mail:juanlook@163.com。通信作者朱小源,E-mail:zhuxy@gdppri.com
(责任编辑 岳梅)
猜你喜欢
食品安全导刊(2022年11期)2022-11-17
现代园艺(2022年9期)2022-06-07
今日农业(2022年4期)2022-06-01
作物学报(2022年6期)2022-04-08
草地学报(2022年3期)2022-03-28
农民致富之友(2020年15期)2020-05-25
江苏农业学报(2019年1期)2019-09-10
农民致富之友(2019年24期)2019-08-20
发明与创新·大科技(2019年5期)2019-07-31
大自然探索(2017年8期)2017-08-28
