
二氧化碳转化为合成气及高附加值产品的研究进展
2022-04-12 03:54邵斌孙哲毅章云潘冯弘康赵开庆胡军刘洪来
化工进展 2022年3期
邵斌,孙哲毅,章云,潘冯弘康,赵开庆,胡军,刘洪来
(华东理工大学化学与分子工程学院,上海 200237)
煤、石油、天然气等化石资源为现代社会发展提供了能源,但日益增长的CO排放已对人类和环境产生严重危害。2018 年,全球CO排放量约为42.1Gt,其中36.6Gt(87%)来源于以化石燃料为基础的能源系统和化学工业。2020年大气中CO浓度已高达416mL/m,是工业革命前的1.45倍。针对现有化石能源体系,碳捕集与封存(CCS)技术是减少CO排放的最有效途径。目前全球CCS工业和示范项目已达到300多个,但总体规模远不能满足社会快速增长的需求。其主要原因在于CCS技术成本高,并且将捕集的CO封存的可持续性和安全性备受质疑。因此,发展碳捕集与利用(CCU)技术,将捕集的CO转化高附加值的化学品和燃料,从而实现CO资源化利用,是一项重要的碳减排技术,对缓解能源危机和实现碳中和具有重要意义。
CO高效转化技术常见产物有碳一化学品(CH、CO、甲醇等)、尿素、乙醇、聚酯、芳烃和烯烃等。CO分子的标准吉布斯自由能(Δ)为-394.38kJ/mol,其固有的热力学稳定性与动力学惰性致使直接合成高附加值化学品效率低、反应条件苛刻、产物收率低。相对而言,CO转化为CO是最切实可行的途径,并且CO 具有高活性,将其与H按一定比例共混可以得到合成气(CO+H),作为平台分子进一步制取液体燃料、低碳烯烃、芳烃等大宗化工和能源高附加值化学品。目前,以CO为原料制备合成气方法包括热催化、电催化、光催化等转化途径,其中热催化转化具有处理量大、效率高、易于大规模工业化应用等优势。相比较热催化过程反应温度高,电催化和光催化还原CO可以在常温常压下进行,也为工业上低能耗绿色生产合成气提供了可行的技术路线。因此将CO热催化、电催化、光催化转化为合成气以及合成气进一步转化高附加值化学品是实现CCU的重要途径(图1),其核心关键是设计和制备高效的催化剂。本文着重对比综述了热催化法、电催化法、光催化3种不同方法将CO高效转化合成气最新进展,总结了合成气进一步转化为高附加值化学品例如烯烃、液态燃料和芳烃等研究工作,分析了各反应过程催化剂设计方法,为CO高效转化利用提供新的思路。

图1 CO2转化合成气以及合成气进一步转化高附加值产品途径
1 二氧化碳转化为合成气
将捕集的CO作为碳源转化为合成气,可实现碳循环利用,在解决环境问题的同时,协同突破资源与能源的碳中和的瓶颈问题。合成气是化学工业生产中的一类重要原料。不同氢碳比(H/CO)的合成气在化学合成中有着不同的应用。合成气的生产主要采用天然气催化重整和煤气化工艺,高度依赖化石原料,造成了环境和能源的双重压力。因此,寻找绿色可替代的合成气生产方法迫在眉睫。利用CO与甲烷或氢气通过热催化,可大规模生产合成气,实现化石能源低碳利用;其次,CO电化学还原与水电解制氢耦合,在常温常压下将其成功转化成不同氢碳比合成气是一种清洁高效的生产方法;最后,利用清洁可再生太阳能通过光催化将CO还原为合成气是一种绿色环保的新方法。
1.1 CO2热催化制备合成气
CO通过催化加氢制取合成气,氢可由传统意义的氢气和富含氢的物质例如甲烷等低碳烷烃提供,通过包括二氧化碳-甲烷干重整(dry reforming of methane,DRM)和逆水煤气变换反应(reverse water-gas shift reaction,RWGS)两种典型反应过程实现。
1.1.1 二氧化碳甲烷干重整(DRM)制合成气
DRM 主要通过CH催化还原CO[式(1)],可生产2倍体积的合成气,是一种有效利用CO的方法。热力学分析表明DRM是一个强吸热反应,高温有利于反应正向进行,但是过高的反应温度对反应装置要求较高,同时利于发生逆水煤气副反应[式(2)],消耗产生的H,造成H/CO体积比下降。更值得注意的是所有合成气生产过程都受到焦化的影响,由C-H-O 三元热力学分析图表明(图2),550~700℃易发生的CO 歧化反应[Boudouar reaction,式(3)]和900℃以上CH的裂解反应[methane cracking,式(4)],都将导致催化剂表面积炭。因此,开发合适的催化剂对提高DRM 反应选择性,抑制催化剂表面积炭意义重大。贵金属催化剂例如Rh、Ru、Ir、Pt、Pd 等活性组分具有反应活性和选择性高、反应温度低、抗积炭和寿命长的特点,但其高成本限制了其大规模应用。而非贵金属催化剂例如Ni、Co、Fe、Mo、Cu 等活性组分具有成本低和储量来源丰富的优势,其中Ni 基催化剂的活性和贵金属催化剂相当,是近年来的研究热点,但Ni 基催化剂在长时间高温条件下容易失活,失活主要原因包括两个:①CH裂解和CO歧化反应引起催化剂表面积炭失活;②金属活性组分Ni高温下烧结,使得催化表面活性点减少。

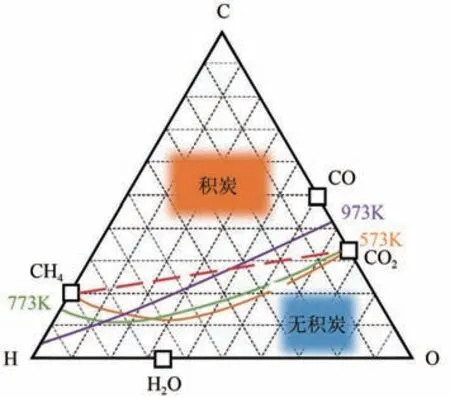
图2 C-H-O三元相图
反应机理的研究对于指导合成高效催化剂具有重要意义,DRM反应机理复杂,通常包含的主要步骤为:①CH和CO的解离或活化;②含有C、H和O元素的中间体在活性位点上的吸附;③表面反应和产物的生成;④CO、H和HO 等产物的脱附。Zhou等利用密度泛函理论(DFT)计算得出在活性Ni(111)表面的反应物、中间体和产物的吸附能以及涉及DRM 过程的可能进行的基元反应活化能(图3),主要反应路径包括CO直接解离出CO 和O,CH解离生成CH或C,CH 和C 被O 氧化,最后CHO 分解生成CO。Zhu 等建立了Ni 催化剂的微观动力学模型,确定了3条反应速率接近的反应路径,其中表面O 氧化C、OH 氧化C 和CH氧化反应路径的贡献率分别为73.1%、8.4%和18.5%,表明C 被表面O 氧化是主要反应途径。因此,碳在催化剂表面沉积的速率和程度由CH解离、CO歧化和表面碳氧化共同决定。Zhang等报道揭示了Mo 在改善传统Ni/Z 催化剂的催化性能和减缓积炭方面起促进作用,存在稳定、高含量的Ni,提供了更有效的甲酸盐中间体的催化途径以及反应过程独特的MoO→ MoCO氧化还原循环,有利于积炭的去除。但由于DRM 反应过程每一步骤还包含多种可能性,目前真正的反应途径和速率决定步骤(RDS)仍存在一定的争议,有待结合催化剂特性深入探讨。
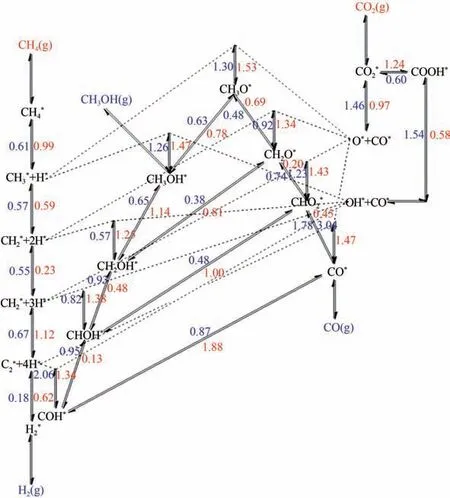
图3 CO2-CH4干重整体系在Ni基催化剂表面的正、逆反应在973.15K的反应能垒[16]
目前普遍认为催化剂的特性与颗粒大小、形貌、金属活性位与载体间相互作用的强弱以及实验条件等因素有关。金属活性组分的尺寸超过临界尺寸(10nm)时,才能满足积炭成核的条件。但在DRM 高温反应条件下,尺寸较小的催化颗粒普遍表现出较差的热稳定性,容易烧结,因此制备高活性、高稳定性、抗积炭催化剂仍然是一个巨大的挑战。Zhang 等设计制备了一种Ni 单原子型催化剂(Ni/HAP-Ce),通过与Ce掺杂的羟基磷灰石(HAP)相互作用,原子级分散的Ni 具有DRM 的高催化活性和抗积炭性能。实验和计算研究表明,孤立的镍原子具有只激活CH中的第1 个C-H 键的独特性质,本质上避免了甲烷深度分解成积炭。由于金属活性组分和载体之间存在普遍的相互作用,通过选择合适的载体调控金属-载体相互作用(MSI),可以提高Ni 在载体表面的分散度,一方面能够提供更多的反应活性位点来提高反应活性,另一方面可以使Ni 纳米颗粒的尺寸小于临界尺寸,从而有效抑制积炭产生,增强催化剂稳定性。Yavuz 等报道了一种设计稳定高效甲烷干法重整催化剂的新策略——单晶边缘纳米催化剂(NOSCE)技术(图4)。单晶MgO 载体的边缘可稳定Mo 掺杂的Ni纳米催化剂(Ni-Mo/MgO),其中Mo掺杂可以增强Ni 的氧化稳定性,促进Ni 颗粒向MgO 晶体的台阶边缘移动。在DRM 反应中具有高效催化活性,并且连续运行850h 以上仍未失活,具有优异的抗结焦和抗烧结性能。通过掺杂另一种金属形成Ni-M双金属合金催化剂也能有效提高热催化DRM 的抗积炭性。Kim 等合成了粒径恒定在5.5nm 左右的NiFe/MgAlO 双金属合金催化剂,与单一金属催化剂相比表现出最佳的DRM 催化活性和稳定性。进一步揭示NiFe 合金表面上Fe 迁移形成了FeO 的关键作用:通过氧化还原机理恢复NiFe 合金并减少碳沉积。

图4 Ni-Mo/MgO催化剂单晶边缘负载纳米催化剂(NOSCE)机制[22]
1.1.2 二氧化碳加氢(RWGS)制合成气
将捕集的CO通过逆水煤气变换[RWGS,式(2)]加氢(尤其是绿氢)还原为合成气,将是未来形成封闭碳循环的重要一步。RWGS 是一个吸热过程,高温有利于反应,但往往伴随着甲烷化副反应[Sabatier reaction,式(5)],导致CO产物的选择性低。因此,开发高温下具有高活性和选择性的催化剂是关键。Kaiser 等分析了在H、CO摩尔比为3∶1的条件下RWGS 反应气态产物的热力学平衡组成(图5)。当反应温度低于600℃时,CH是主要产物;超过700℃时,CO产物明显增多。但从降低能耗和成本角度分析,应避免高温反应,因此需对RWGS反应机理深入理解,特别是结合实际催化剂探讨反应路径和反应动力学,从而获得催化剂设计与制备规律,具有重要意义。
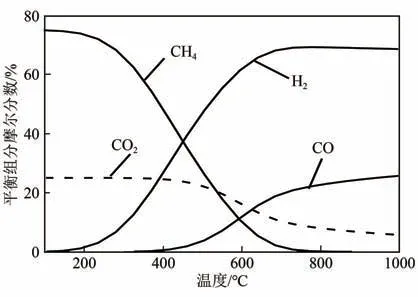
图5 RWGS反应气态产物的热力学平衡组成
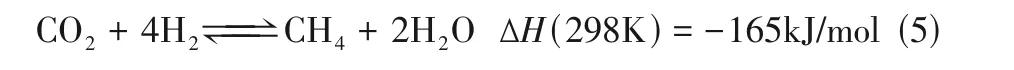
近年来,结合多种原位表征技术、同位素示踪方法和基于DFT 计算对催化剂表面吸附物种演化的跟踪,对RWGS 反应机理的研究取得了重大进展。表1列出了RWGS反应遵循的两种典型反应路径:①氧化还原机理,即CO在催化剂表面解离为CO和O,H与O反应,但并不直接参与CO的还原;②中间产物机理,即H与CO在催化剂表面直接反应生成中间体(如甲酸盐、羧酸盐或碳酸氢盐等),然后进一步加氢生成CO和HO。氧化还原机理认为在金属纳米颗粒表面形成的CO 的结合强度可以决定产物的选择性。CO与纳米颗粒的强相互作用导致C—O键解离,有利于CH的形成,而CO与金属的弱相互作用有利于CO 本身作为产品。因此,调控金属表面对CO 吸附强度可以大幅提高逆水煤气反应产物CO 的选择性。中间体产物机理认为,中间物只在载体与金属纳米粒子的界面上形成,金属纳米粒子上吸附的氢原子可以与中间产物反应,以CO和HO的形式解吸。Szanyi等报道了两种Pd/AlO负载型催化剂,基于原位红外-质谱表征发现CO首先与氧化铝上的羟基结合生成碳酸氢盐,Pd上吸附的H与之反应生成甲酸盐中间体。由于Pd有优先生成CO或CH的两组不同晶面,与CO弱相互作用的Pd位点解吸产生CO,与CO强相互作用的Pd位点,CO进一步加氢生成CH。
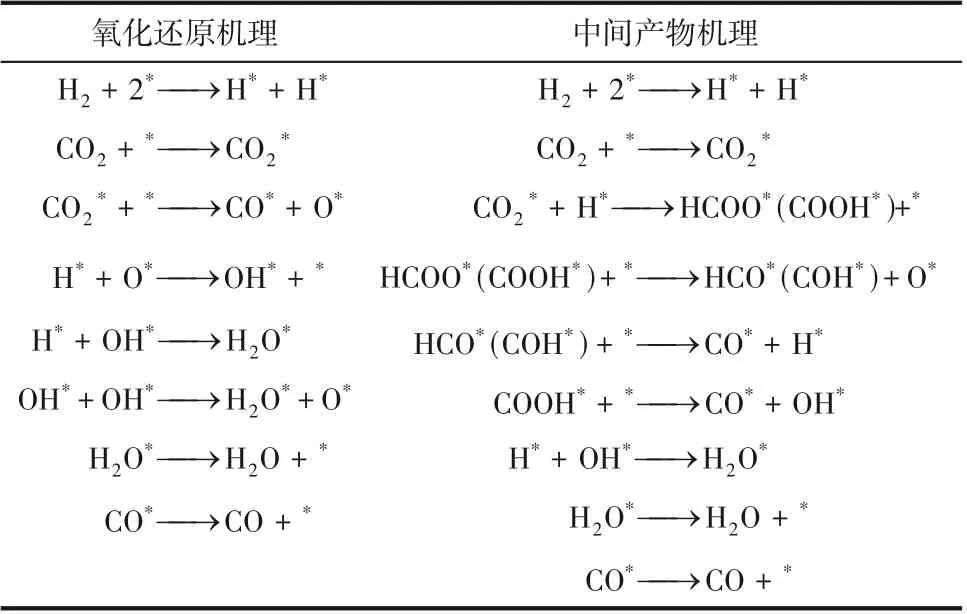
表1 RWGS反应的氧化还原机理和中间产物机理[25]
同时,催化剂表面的空位也被认为有助于CO的吸附和活化。因此,氧空位浓度高、金属-中间物种键能强度弱的催化剂对RWGS反应具有较好的催化效果。最近Willauer 等开发了一种低成本、高活性和高选择性的K-MoC/g-AlO催化剂,450℃时CO转化率为44%,CO 选择性为98%,运行10 天内没有任何失活迹象。通过DFT 计算进一步发现反应过程中有更多氧覆盖的MoC表面对CO吸附增强,容易加氢转化为CH,会导致CO 选择性下降。Hu等针对乙烯工业过程裂解高温烟气,将钙循环(calcium-looping,CaL)和RWGS 反应相结合,通过设计氧化还原异质结的Fe/Co 合金催化剂(图6),在同一固定床中成功实现了长周期高温CO捕集和原位转化,CO转化率高达90%,CO选择性100%。

图6 双金属催化剂Fe5Co5Mg10CaO异质结-氧化还原机理[30]
1.1.3 CO热催化制合成气的工业化进展
以二氧化碳规模化利用技术为核心的工业化碳负排方案中,CO-CH通过DRM 制合成气是一种绿色的生产方式。目前天然气除作为燃料外,约15%用作化工原料,假设其中30%用于CO-CH干重整并耦合合成气直接制烯烃,则各类烯烃产量可达740 万吨,同时直接消耗CO约1020 万吨。2010年12月,美国Carbon Sciences Inc.公司与加拿大萨斯喀彻温大学(UOS)就DRM 催化剂技术签署了全球独家许可协议。在反应器连续2000h的实验中,UOS开发的催化剂转化率达到92%,无明显的烧结和积炭现象。国内上海高等研究院孙予罕教授团队在成功解决Ni-CaO-ZrO纳米复合催化剂的抗焦化问题基础上,完成了催化剂研制和反应器模拟研究以及百吨级催化剂的工程放大和生产,并于2017 年在山西潞安集团建成了全球首套万吨级CO自热重整制合成气中试装置,日转化利用CO高达60t,合成气产量高达20m/d。该CO-CH干重整制备合成气技术的成本为500~600CNY/t 合成气,与煤制合成气技术的成本相当,相较于目前传统的水蒸气重整生产合成气,其成本可以降低20%。实际DRM工业化应用过程中仍需解决的问题包括:①高压操作的必要性,尤其是生产得到的高压合成气对于进一步转化为其他产品具有优势;②调控合成气中碳氢比,获得目标合成气。
虽然RWGS反应是活化CO的重要的一步,但目前还未见CO转化利用的工业开发应用。Rezaei等通过Aspen 模拟分别评价了RWGS 工艺和DMR 工艺两种不同合成气生产方式的经济性。与RWGS工艺相比,DMR 工艺生产H/CO 比为1 和2 的合成气的总成本更低。以H价格、CH价格和碳税进行盈亏平衡分析发现当H价格为1000USD/t,CH价格和碳税分别为740USD/t 和44USD/t 时,RWGS 工艺与DMR 工艺相当。目前的研究重点是降低H生产成本和强化RWGS反应,从而降低工业应用RWGS过程的总成本。但是RWGS工艺作为合成高附加值产品串联反应的第1步,在CAMERE甲醇合成工艺中得到了实际工业应用。同时,RWGS过程与烷烃脱氢耦合是工业化高效生产烯烃和合成气的途径。因此,通过RWGS 反应将CO转化为高附加值化学品和燃料具有广阔前景。
1.2 CO2电催化转化合成气
相比较于热催化转化CO,电催化还原CO的方法具有催化效率高、操作条件简单和反应条件温和等特点。目前大部分已报道的电催化剂都集中于CO高选择性催化转化为CO。本节通过概述最新的高效CO电催化转化合成气的催化剂研究进展,探讨其工业化应用前景。
1.2.1 电催化还原CO基本原理
CO电催化还原为CO的反应机理主要包括3个步骤:①CO的活化生成CO;②经过质子-电子转移(PET)先生成COOH中间体,再进一步生成CO和HO;③吸附在催化剂表面的CO脱附。由于析氢反应电位低,是溶液体系CO电催化还原不可避免的副反应。由反应机理分析可知,相应的高效电催化剂应满足:①对CO具有较强的吸附活性和还原能力;②对关键中间体(COOH)具有适当的吸附强度;③CO吸附能适中、还原能力弱。因此,开发性能优异、合成方法简单、来源丰富和低价的CO电还原催化剂是关键。
1.2.2 电催化新材料
目前高效CO电催化剂主要包括纳米单金属催化剂、纳米合金催化剂以及过渡金属单原子催化剂等。建立电催化剂微观几何结构与电子分布的调控方法,探索微观结构与催化性能的关系是研究的重点。微观几何结构中催化剂的尺寸效应尤为显著。Liu等通过DFT计算发现,当Cu纳米颗粒降低至由309个原子组成时呈现显著的尺寸效应。当颗粒由147 个原子组成时,CO电催化转化为CO过程具有最合适的COOH吸附能以及CO脱附能,对应最低的CO 生成能垒,同时也能抑制H的形成。类似的尺寸效应在超细Au 纳米颗粒也有体现(图7)。
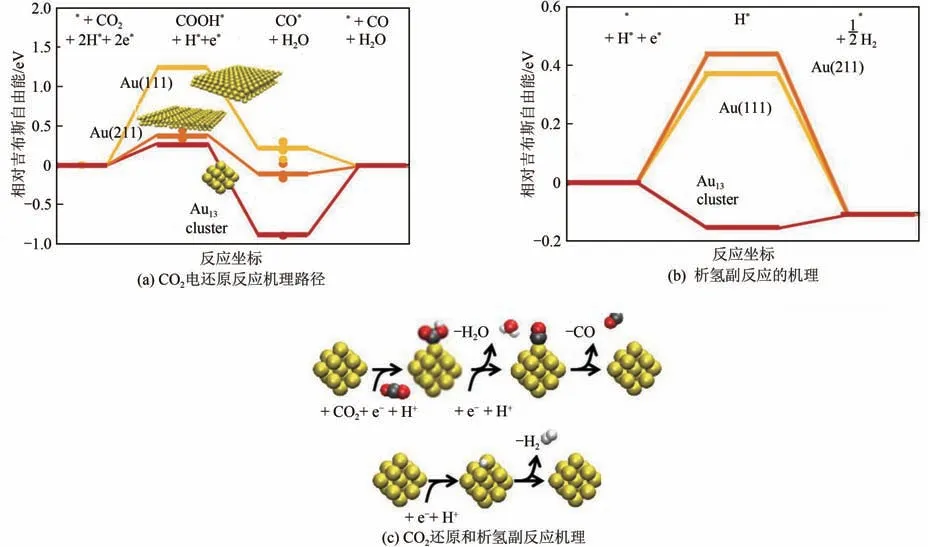
图7 Au纳米颗粒尺寸效应对电催化还原CO2机理的影响[39]
进一步将催化剂金属活性组分尺寸降低到原子尺度,金属单原子电催化剂呈现出不同寻常的CO电催化还原活性,并且金属与载体之间的MSI相互作用更为显著。这种效应往往能够显著调控电催化反应中各个中间物种的吸附能,从而提高反应的活性。因此,单原子金属电催化剂的原子利用率极高,催化位点明确,易于剖析催化反应路径等优点。其中Ni单原子催化剂表现出极佳CO电催化还原性能,CO 的法拉第效率几乎可达到100%。Gong 等利用MOF 中低沸点金属Mg,通过热解法控制Ni 单原子的N 配位数Ni-N-C(=2,3,4),其中Ni-N-C 的CO 法拉第效率最高达到97%,对应的转化频率(TOF)高达1622h。此外,其他过渡金属单原子电催化剂如Fe、Co 等,均能够通过适当的合成方法精确调控配位环境从而实现CO高效转化为合成气。载体的性质也会对催化活性起到至关重要的作用,Zhu 等通过DFT 计算发现石墨烯上的Fe-N位点对CO的吸附能过强而易被毒化,而富缺陷的石墨烯则能够促进Fe-N位点上CO 的脱附从而提高CO的法拉第效率。
通过优选双(多)金属掺杂组成和配比精准调控催化活性位点d带电子结构的策略,也广泛应用于提高CO电催化效率,调控合成气的碳氢比。Han 等报道了一种Cu-Co 合金电催化剂,其在含离子液体的甲腈电解液中CO 的法拉第效率最高可97.4%,Cu和Co的比例决定了合成气中H和CO的比例。此外,He等也发现Co-N位点能够高效析氢而Ni-N位点能够高效产出CO,两者的共存能够实现合成气比例的精准调控。
结合金属单原子催化效率和双金属催化的调控性,金属双原子对电催化剂协同促进催化的现象引起了关注。Zhao等制备了含有丰富Ni-Fe双原子对位点的电催化剂,在很宽的电势范围(-0.4~-1.0V)内均能高效产出比例为8∶1的合成气,CO的最优法拉第效率高达98%。DFT计算进一步揭示了这种独特Ni-Fe 双原子对可以使CO在两个金属位点交替协同促进转化为CO(图8)。
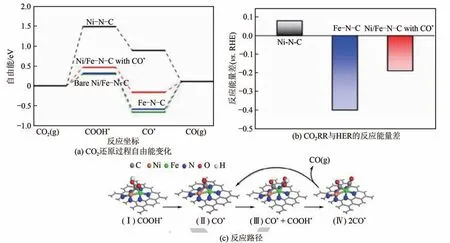
图8 Fe-Ni双原子对位点上CO2电催化还原的机理[47]
1.2.3 CO电催化制合成气的工业化进展
近年来电催化反应体系从实验室小试、中试示范到工业应用不断发展,电极从最初的玻碳电极变为气体扩散电极;电解池类型从H型电解池到流动池,再到兼具两者优点的膜电极体系(MEA),目前工业化的CO电解电流密度可在0~1000mA/cm范围内变化,对应的合成气比例在(4∶1)~(1∶1)灵活调控。
近日,中国石油和化学工业联合会在内蒙古对CO电解制合成气中试装置进行了现场考核。此装置自从2020年8月开车以来,一直保持稳定运行状态,其能够稳定产出碳氢比为0.52∶1 的合成气,直流电耗电量为6.69kW·h/m。此装置每年可处理30t CO,生产45000m合成气,副产22500mO。因此,CO电催化还原生产合成气是极具前景的节能减排策略。
据CO电催化还原成本分析(图9),目前电催化还原CO制CO 成本为130USD/t,仅次于甲酸(80USD/t)。但是液态的甲酸需从电解液中分离提纯,其对应的成本高达60USD/t。而气态CO 可采用简单的变压吸附进行分离提纯,对应成本则仅为10USD/t。因此,CO电催化制合成气最具经济性,并且合成气可以作为重要化工平台气原料,制备系列的精细化学品,进一步提高其附加值;同时在技术层面最易实现大规模电解技术。
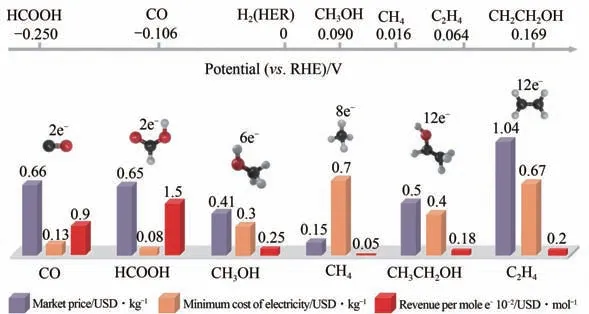
图9 CO2电催化还原制备各种产物的经济效益分析[52]
但目前工业化电解CO制合成气还面临着巨大的挑战,主要体现于:①工业化过程需施加较高的电压提高反应速率,确保连续规模化生产量。如何在高电流密度下(>200mA/cm)提高CO 产物的选择性,从而避免后续产物的分离是重要问题。②在电催化过程中,二氧化碳还原(CORR)和水还原(HER)反应具有竞争关系,电解过程既要保持两个反应的活性,又要同时实现碳氢比可调的合成气,具有一定的挑战性。③阴极CO电催化还原效率还会受到阳极半反应(通常为析氧反应)的影响,整个电解体系中阳极上的电能损耗可高达90%。如何提高电解整体能源效率,需兼顾解决电催化析氧反应的动力学制约问题。
1.3 CO2光催化转化合成气
光催化技术利用清洁可再生太阳能转化为化学能,兼顾能源、环境和经济要求,是最具前景的绿色转化技术。本节聚焦光催化CO制合成气的机理,通过概述光催化最新材料及改性策略等研究动态,以期探讨规模化工业生产前景。
1.3.1 CO光催化转化机理
CO光催化转化是一个典型的多电子转移过程,包括3个步骤:光催化剂对光的吸收;光生载流子的产生、分离和传输以及光生载流子和反应物之间的化学过程[图10(a)]。光合作用是最经典的CO光催化原理,通过叶绿体,利用光能将CO和HO转化成储存着能量的有机物,并且释放出氧气的过程。光合作用过程至少包括一个光能转换的叶绿素分子(P)、一个原初电子受体(A)和一个原初电子供体(D),才能将光能转换为化学能,CO得到原初电子受体A提供的电子而还原为葡萄糖等有机物。光合作用过程中CO并没有直接参与原初电子传递,其原因在于CO稳定性高、还原电位高,与叶绿素的带隙能不匹配。为了寻找高效CO还原光催化剂,分析对比常见的半导体光催化剂的带隙能与CO不同还原产物的电极电位,如图10(b)所示,光催化剂的导带需高于-0.52eV,才能将CO还原为CO。同时由于析氢反应的还原电位略低于CO/CO电位,因此析氢反应在CO转化制合成气过程中不可避免。要提高CO的光催化效率,光催化剂的结构调控是关键。

图10 光催化CO2转化的过程和不同物质的氧化还原电位与光催化剂的带隙[57-58]
1.3.2 多孔光催化新材料
对于气固光催化过程,CO气体吸附在固体光催化剂表面是首要步骤。多孔光催化材料例如多孔聚合(POP)、金属有机框架材料(MOF)、共价有机框架材料(COF)等新材料兼具CO吸附富集和光催化转化双功能,越来越受到关注。
POPs 是一类新型的结构多样的高比表面聚合物材料,通过结构单元的设计与调控,强可见光吸收活性、光电性能可调以及高化学和热稳定性等优点。近期,Yang等开发了曙红Y基共轭多孔聚合物,在无任何光敏化剂或牺牲剂存在下,能有效地将CO和HO光催化还原为合成气。
MOFs 中存在的开放金属位点既可以作为CO分子的吸附位点,也可以作为CO光催化还原活性位点,各种MOFs 材料如HKUST-1、MIL-101、MAF-X27-OH、MOF-525-Co、NNU-31-M 等已成功应用于CO光催化还原。目前,MOFs 金属节点物种、配体结构及其形成的晶体框架结构与光催化性能的关系是研究的重点。Dong 等通过调节MOF(PCN-250-Fe3)金属簇节点中Fe的价态,实现了CO转化活性的大幅提高,同时在光催化反应系统中可以检测到H产物生成,形成合成气。
COFs材料中存在大量π共轭结构,可使电子离域,已被用作电荷载流子传输的优良介质和光催化剂,并且COFs 表现出优异的CO吸附能力,近年来,D-A基COFs的探索也受到了广泛关注。Lu等构筑了新型卟啉-四硫富瓦烯COF,缺电子金属卟啉(TAPP)络合物和富电子四硫富瓦烯(TTF)的链接增强了电荷分离效率、改善了光吸收,使CO高选择性还原为CO。Zou 等合成了2,2’-联吡啶基COF,与Ni 配位得到单原子分散(Ni-TpBpy),在将CO光催化还原为CO 同时产生H获得合成气(图11)。
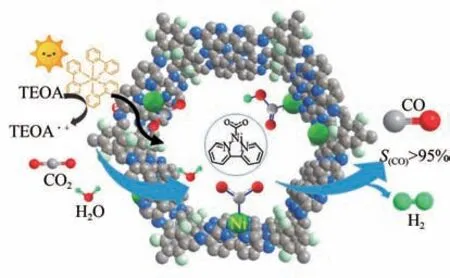
图11 Ni-TpBpy光催化选择性还原CO2[64]
1.3.3 光催化剂的改性策略
光催化还原CO和HO 被认为是一种绿色和可持续的合成气生产方法。然而,该光催化反应的效率、合成气中碳氢比的调控还存在较大的挑战。最新的光催化改性策略聚焦于异质结的构筑、空缺位缺陷的调控等,以促进与调控CO还原反应(CORR)和析氢反应(HER)。
合理构建异质结构能够有效提高光催化剂内部载流子的分离效率,并提升氧化还原能力。Yu等将Co 活性中心与三嗪框架(CTF)配位,形成分子内异质结,光吸收明显增强,能够在10h内产生3303μmol/g 的合成气(CO∶H=1.4∶1),产率是不含异质结的CTF 的3 倍。Song 等通过静电自组装形成系列层状双氢氧化物/MoS异质结构的复合光催化剂,在可见光照射下将CO光还原得到的合成气,碳氢比从(1∶1)~(9∶1)可调(图12)。最近,Hu和Li等报道了一种环境稳定型黑磷异质结复合材料Pt/BP-OvMBWO,用于光催化CO还原制备合成气[CO∶H=(1∶1)~(2∶1)],CO 和H的最大生成速率分别高达20.5μmol/(g·h)和16.8μmol/(g·h),且具有优异的循环使用稳定性。
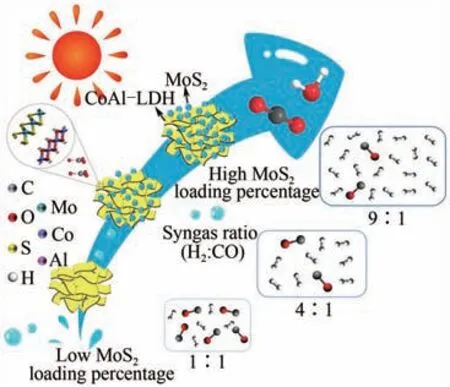
图12 LDH/MoS2纳米复合物通过CO2光还原合成可调谐合成气[66]
将光敏剂与催化剂偶联也是构筑异质结最常用方法。Zheng 等以超薄的层状双氢氧化物(LDH)为载体制备了高度分散Pd 的催化剂,与Ru 络合物敏化剂配合形成高效异质结,在可见光照射下得到的合成气碳氢比在(1∶0.74)~(1∶3)可调。Wu 等在氮掺杂碳上构建Mn 单原子催化剂MnSAs。以[Ru(bpy)]Cl作为光敏剂,以三乙醇胺作为牺牲剂,光催化CO还原制合成气。CO和H的产气速率分别高达1470μmol/(g·h)和1310μmol/(g·h),碳氢比从1.12~0.43 可调。同时,光催化剂的缺陷通常可以产生更多的电子空位,以强化电子空穴的分离,并增加CO的吸附。Wang等通过构建富含S空位的VS-ZnInS纳米片,强化光吸收、电子-空穴分离和CO吸附,同步促进CORR 和HER 反应,CO∶H比为1∶1。与纯ZnInS相比,合成气产率提高了约4.73倍。Yang等报道了一种在聚合氮化碳(PCN)光催化剂表面生成氮空位(NV)的方法,以加速PCN光生载流子的分离和转移动力学。在可见光下,NVs-PCN 的合成气产率几乎是原PCN的10倍。重要的是,通过调节NVs的浓度,合成气H∶CO比可以在(0.24∶1)~(6.8∶1)之间调节。
1.3.4 光催化制合成气规模化研究的挑战
目前,光催化还原CO的研究主要集中在实验室规模,距离规模化实际应用还面临诸多问题,例如:①对CO光催化还原的机理,包括具体中间体、反应途径和反应动力学认识不足,对提高CO光催化还原效率还处于“试错”阶段。②实验室研究将固体光催化剂粉末直接加入CO溶液中,以充分提高光吸收、气固两相接触和光催化效率。但是悬浮体系伴随着光催化剂的后续分离和回收难度大、重复利用效率低等问题。③目前CO光还原产物主要集中于C化合物,通过光催化还原CO获得高附加值产物仍面临挑战。因此,开发具有高活性、高反应选择性和稳定性的新型光催化剂迫在眉睫。合理设计高效的无贵金属助催化剂,有助于构建低成本。高效的光反应器和反应系统工程对于实现高效的CO减排至关重要。
光催化剂成型工程技术研究是工业化应用的首要关键,催化剂固定化避免了光催化剂分离问题,是一个值得关注的新方向。采用气固相反应技术可实现高效气固光催化,避免水溶液悬浮体系中CO气体溶解度低、光催化剂相容性差、负载型颗粒易脱落等稳定性问题。通过反应器设计改进紫外光源穿透模式,可以较大程度提高光催化效率,使光催化制合成气具有潜在的工业化应用前景。
2 合成气制高附加值产品
CO通过热催化、电催化和光催化过程可高效转化为合成气,合成气作为生产合成氨、羰基合成醇、费托合成的原料平台,可进一步转化成高附加值化学产品和燃料,从而提升整个CO减排过程的可行性和经济性,具有重要的研究意义。本节聚焦合成气制低碳烯烃和芳烃的最新进展。
2.1 合成气制低碳烯烃和液体燃料
合成气制长链烃的催化过程为经典的费托合成(Fischer-Tropsch synthesis,FTS)。如图13 所示,其主要反应机理包括:首先CO和H分子在催化剂表面发生解离吸附;然后CO 解离形成的C 物种加氢形成CH中间体(0≤≤3);这些中间体会进一步发生C—C偶联生成CH中间体或者生成CH;最后CH经加氢或脱氢形成不同碳数的烷烃和烯烃。因为费托合成遵循表面偶联机理,产物服从Anderson-Shulz-Flory(ASF)分布,其中产物C~C烃类的选择性不超过58%,汽油(C~C)组分选择性不超过48%。因此,如何将合成气高选择性地定向转化为特定的高附加值化学品具有重大挑战。
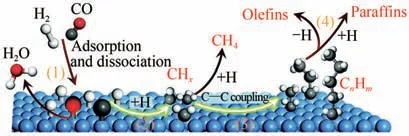
图13 费托合成反应机理中关键步骤[72]
目前费托合成的研究主要集中于金属氧化物及其碳化物型催化剂,例如Fe、Co和Ru等。Sun等研制出棱镜型的CoC催化剂,可以在250℃、0.1MPa温和的条件下高选择性地将合成气催化转化为轻质烯烃,选择性为60.8%,而甲烷选择性低至5.0%。近期,Ding等发展了一种新型疏水性FeMn@Si催化剂,在模拟工业反应条件下(320℃和2~3MPa),实现了合成气高选择性制取烯烃,烯烃产率高达36%,选择性可达65%,CH和CO等C副产物选择性显著降低至22.5%。进一步研究发现,FeMn@Si表面的SiO疏水壳层可保护碳化铁活性相免受水的氧化,并保持碳化铁活性相处于良好的稳定状态。Ma 等开发出Na 修饰的FeC-ZnO 催化剂,在340℃、2MPa的条件下烯烃选择性高达78%,其中主要以高碳-烯烃为主,烯烃的时空收率超过4000mg/(g·h),同时甲烷和CO的选择性分别控制在8%和25%。
包信和院士团队提出了“OX-ZEO”一步法制取低碳烯烃技术路线,如图14 所示,其反应机理为合成气先在金属氧化物上反应生成乙烯酮(CHCO)中间体,该中间体进入邻近的分子筛孔道,在分子筛择形酸性位催化作用下生成低碳烯烃。他们采用ZnCrO金属氧化物和介孔SAPO-34(MSAPO)分子筛组成双功能催化剂,具有优异的低碳烯烃选择性,在400℃、2.5MPa、合成气H/CO为1.5 反应条件下,低碳烯烃选择性高达80%,突破了ASF 分布,但CO 转化率较低为17%。基于此反应,2019年9月,中国科学院大连物理化学研究所与陕西延长石油集团合作,成功完成煤经合成气直接制低碳烯烃技术工业试验,CO 单程转化率超过50%,低碳烯烃选择性优于75%,总体催化性能优于实验室水平,进一步验证了该技术路线的先进性和可行性。最近,包信和院士团队通过对比研究了ZnCrO-SAPO-34 和MnO-SAPO-34 两种双功能催化剂,进一步揭示了金属迁移的影响,Zn物种易于迁移至SAPO-34 上,形成Zn-OH,屏蔽了分子筛的Brønsted酸中心,导致低碳烯烃选择性下降。相比之下,Mn物种不会发生金属迁移现象,在纳米尺度上MnO与SAPO-34 两种催化活性位越近,越有利于乙烯酮中间物扩散,低碳烯烃选择性越高,这为进一步开发该类双功能催化剂提供了基础。Wang 等开发了由ZnO-ZrO双金属氧化物和SAPO-34 分子筛组成的双功能催化剂。在400℃、1MPa、H/CO=2反应条件下,低碳烯烃选择性高达74%,但CO 转化率为11%。他们通过改进分子筛结构得到ZnO-ZrO/SSZ-13 的双功能催化剂,将CO转化率提升至29%,同时低碳烯烃选择性77%。机理研究表明,CO首先在ZnO-ZrO表面转化为甲酸类化合物,然后甲酸类化合物通过氢化反应转化为甲氧基化合物。最后甲氧基化合物经分子筛进一步催化为烯烃。

图14 在ZnCrOx/SAPO-34催化剂上合成气经乙烯酮中间体转化为低碳烯烃的反应机理[72]
此外,合成气也可以通过费托合成与加氢裂解/异构化反应耦合而成的接力催化方式高选择性地转化为液体燃料如汽油和异构烷烃等。Sun 等通过无溶剂研磨法制备出弱酸性Silicalite-1 分子筛封装金属钴催化剂。在CO 转化率约为30%的情况下,汽油选择性高达70%,异构烷烃选择性可达30.7%。包信和院士团队开发出ZnMnO/SAPO-11 双功能催化剂,该催化剂可以直接选择性将合成气转化为高品质汽油。在CO 转化率为20.3%时,汽油在烃类中的选择性高达76.7%,CH的选择性仅为2.3%。刘中民院士团队构建出CuZnAl 与AlO混合组分和ZSM-5组成的双功能催化剂,实现了合成气向汽油的高效转化,CO 单程转化率高达86.3%,除开CO后C~C汽油产物选择性高达80.6%,并且催化剂在110h的催化测试中催化性能保持稳定。此外,C含氧化合物特别是乙醇可用作燃料添加剂和绿色有机溶剂。Wang 等设计出由K-ZnO-ZrO、H-MOR 和Pt-Sn/SiC 组成的三步接力催化体系,通过将合成气制甲醇、甲醇羰基化制乙酸、乙酸加氢制乙醇3个反应按接力催化的方式进行高效集成,实现了合成气一步高选择性制乙醇。乙醇选择性可达70%~90%,突破了传统过程的乙醇选择性极限值。
2.2 合成气制芳烃
通常合成气制芳烃路线分为两步:先把合成气转化为甲醇,再通过甲醇芳构化反应得到芳烃。但两步法工艺繁琐且甲醇制芳烃催化剂积炭失活严重。并且由于受费托合成反应产物ASF 分布限制,产物选择性低。近年来,接力催化策略(OXZEO)在合成气转化中展现出独特的优势:通过反应的耦合不仅强化了反应过程,还保证了目标产物的高选择性合成。因此,将合成气制甲醇催化剂与H-ZSM-5 耦合,实现了合成气直接高选择性制芳烃。Ma 等将高效制备-烯烃的Na-Zn-FeC与改性处理后的介孔H-ZSM-5 分子筛混合,在340℃、2MPa 条件下实现了CO 转化率为89%,芳烃选择性为51%,时空收率可以达到16.8g/(g·h),同时CH和CO选择性分别抑制在10%和27%的优异性能。Wang 等将ZnO-ZrO双金属氧化物和H-ZSM-5 分子筛组成双功能催化剂,在400℃、3.0MPa、H/CO=2 的反应条件下,CO 转化率可达20%,芳烃选择性高达80%,甲烷选择性抑制低于3%。更为重要的是该双功能催化剂在1000h 反应过程中仍可保持稳定。最近,谢在库院士团队将CrO定向分布于ZSM-5 分子筛的(100)和(101)特定晶面得到高效双功能催化剂,因其CO加氢形成的C中间体易扩散至邻近的ZSM-5分子筛孔道内酸性位点发生芳构化反应,在395℃、7.0MPa、H/CO=1、1L/(g·h)反应条件下,CO转化率高达49.4%。
但是合成气制芳烃双功能催化剂也存在一些问题:目前报道的CO 单程转化率大部分不超过50%,较传统的费托合成催化剂反应活性普遍偏低。因此需要进一步开发出高活性的多金属氧化物来提高催化剂的反应活性。此外,接力催化反应条件一般发生在400℃左右,升高温度对水煤气反应(WGS)有促进作用,导致产生CO副产物,选择性甚至高达43%。未来还需要进行更深的反应机理以及动力学研究,从催化原理本质上抑制WGS反应发生。
3 结语与展望
将CO高效转换为合成气,进一步通过费托合成路线或接力催化路线转化为高附加值化学品和燃料成为发展高效利用非油基资源的新途径(图1)。基于热催化、电催化和光催化这3 种不同方法以CO为原料制备合成气的研究中已经取得了显著进展,但距离大规模的工业化应用仍存在较大挑战,开发出高效、高活性、低成本且稳定的催化剂是各技术推广应用的关键。本文对比和总结了目前热催化、电催化、光催化3种利用CO制备合成气技术,见表2。其中应用成熟度最高的是加氢技术和甲烷干重整等热催化技术,但反应条件苛刻、过程能耗大和催化剂稳定性等问题仍亟待解决。未来需要不断完善催化剂活性位识别-结构-性能三者关系,实现耐高温抗积炭高效催化剂的规模化制备;其次,通过整体系统的内部热能利用和系统强化或耦合外部新能源系统供能,解决并实现反应器优化及其与反应过程的能量匹配;最后,利用合成气作为后续化工生产的基础原料制备高附加值化学品,形成完整集成工艺产业链,实现二氧化碳热催化制备合成气的规模化应用是其重要方向。

表2 CO2热催化、电催化和光催化三种技术方法制备合成气比较
随着光伏、风电等绿色电源的发展,光、电催化将作为一种更加绿色高效的技术用于CO转化制备合成气。由于电催化剂在合成气生产过程中容易出现CO中毒、高压下H气泡的抑制和活性位点的减少等失活问题,限制了该技术的大规模应用,因此未来在设计高效合成气生产催化剂的过程中应考虑以下方面:①在催化剂表面形成多孔结构,促进气体扩散;②优化催化材料的表面亲疏水性,平衡水和CO的吸附;③合理设计催化剂独特的多相界面,防止CO 中毒,增加反应活性位点数量。直接利用太阳能和地球上丰富的原料(HO、CO)光催化生产合成气为CO资源化以及绿色能源利用提供了全新途径。光催化剂活性主要受到电荷分离和转移的制约,合理设计和制造高效且稳定的光催化剂是光催化CO产合成气过程的核心。通过元素掺杂、缺陷工程、贵金属负载和异质结构筑等策略,可显著提高电荷载流子分离效率。基于光催化过程电子和质子转移途径以及化学键的形成/断裂等机制的深入剖析,突破性提高光催化剂稳定性、转化率和产率是光催化规模化应用方面的关键。
本文重点阐述了CO高效转化为合成气的途径,未来将CO捕集与转化过程耦合是CCU研究的重要研究方向,通过设计开发兼具CO吸附捕集与CO催化转化的双功能材料,有望实现CO高效捕集与原位转化成高附加值产品一体化技术。充分提高能源利用率、有效降低减排成本,解决CO捕获后提纯和运输的高额费用以及埋存所带来的安全隐患,从而提升整体CCU技术的可行性和经济性。
猜你喜欢
科学家(2022年4期)2022-05-10
疯狂英语·新阅版(2021年9期)2021-10-30
第一财经(2019年8期)2019-08-26
中国科技纵横(2019年3期)2019-03-25
分析化学(2018年4期)2018-11-02
分析化学(2017年12期)2017-12-25
作文·初中版(2017年6期)2017-06-16
未来英才(2016年14期)2017-01-12
分析化学(2015年3期)2015-04-20
