
面向临床研究的基因测序项目的设计原则、管理流程与质量控制标准
2022-04-06 12:36刘阳许喆程丝石延枫林金嬉孟霞姜勇李昊
中国卒中杂志 2022年3期
刘阳,许喆,程丝,石延枫,林金嬉,孟霞,姜勇,李昊
基因检测是对受试者的DNA进行检测以寻找可能的遗传变异的过程。高通量测序是一种基因检测技术,能够对数百万个DNA片段进行平行序列测定,因此可以同时对大量遗传变异进行鉴定和分析。在临床研究中引入基因测序,可以将受试者的遗传变异与临床表型、生物标志物等数据相结合,为疾病机制的研究和药物靶标的开发提供丰富的资源[1]。临床研究的样本收集时间跨度往往以年为单位,一些大型研究还涉及多中心管理,因此面向临床研究的基因测序项目对管理流程、分析手段及质量控制的标准化程度要求很高,需要与之相适应的项目管理框架。本研究旨在确定面向临床研究的基因测序项目的设计原则,搭建项目管理与质控标准化流程,并将该流程应用于一项全国性多中心大型临床研究,从而确定质量控制标准。
1 方法
1.1 基因测序项目框架的确定
1.1.1 文献复习 通过文献复习学习生物医学研究中的基因测序数据分析流程[2-5],调研国际、国内大规模测序队列的建设现状[6-9],制订面向临床研究的基因测序数据处理流程。
1.1.2 专家咨询 遴选国内基因组学、生物信息学、统计学、生物样本库、临床试验、数据管理等领域的专家,调研面向临床研究的基因测序项目的需求,完善基因测序项目设计原则并确定管理规范。
1.2 基因测序项目的设计原则 通过整理吸纳文献复习结果和专家咨询意见,面向临床研究的基因测序项目需要能满足可扩展性、可重复性及可溯源性的需求。针对这3点需求,分别确定了如下设计原则:
1.2.1 可扩展性 随着对疾病机制研究的深入,越来越多疾病相关的遗传变异被发现。在进行临床研究基因检测的设计时,如果仅包含当时所了解的疾病相关遗传变异,一方面无法涵盖今后可能报道的新变异,另一方面也很难进行探索性的挖掘。全基因组测序(whole genome sequencing,WGS)是对受试者的所有DNA进行的高通量测序,可以覆盖和识别整个基因组的变异,包括单核苷酸变异(single nucleotide variant,SNV)、短插入/缺失变异(insertion-deletions,INDEL)以及更长的结构变异,能够满足临床基因研究的可扩展性需求。
1.2.2 可重复性 科学研究的统计功效需要有一定的样本量,而高通量测序技术依赖复杂的试剂、硬件和训练有素的人员[10],一个批次所能测定的样本有限,因此临床研究的基因测序项目通常涉及多个批次。批次效应(batch effect)是指样本之间的变化不是来自真实的生物学差异,而是来自实验或技术之间的差异[11-12]。批次效应会降低研究结果的可重复性,导致假阳性和假阴性关联,甚至可能会产生误导性生物学或临床结论[13]。解决批次效应,依赖于全面的研究项目设计、可靠的质量控制方案、仔细的执行过程记录,以及恰当的统计建模方法,这些在临床研究基因测序项目的方案设计中都应该考虑到。
1.2.3 可溯源性 研究人员进行大规模临床研究的课题分析时可能会发现感兴趣的现象需要回溯到单独的样本,这就对基因测序数据的可溯源性提出了要求。临床研究往往涉及多中心、长时间的样本收集,对于基因测序项目,一方面是管理上的挑战,另一方面也是机遇,因为遗传数据本身能够对样本质量和来源形成反馈。在基因测序项目管理流程的搭建中,应当充分利用这一点。
1.3 基因测序项目管理流程的应用 数据处理流程搭建好后,需要在真实的临床研究中确定合适的质控标准。中国国家卒中登记Ⅲ(China national stroke registry-Ⅲ,CNSR-Ⅲ)队列是住院的急性缺血性脑血管事件患者的全国前瞻性登记研究,共有15 166例缺血性卒中患者或TIA患者,涉及201家医院,是一个典型的多中心队列[14]。CNSR-Ⅲ研究设计了遗传亚组并进行了全基因组测序,是非常好的基因测序项目管理流程应用场景,因此本研究将在CNSR-Ⅲ队列上实施并确定质控标准。
2 结果
2.1 样本处理
2.1.1 样本类型 遗传研究的目标是检测患者的生殖系基因变异,因此理论上来说,来源于患者的任何细胞都含有同样的变异,都可以用于基因测序[4]。为了取样方便,临床研究中通常保留患者的外周血白细胞用于基因测序。采集血样标本需要送到指定实验室分离血清、血浆和白细胞,随后进行中心化储存和基因测序安排。在CNSR-Ⅲ研究中,预先确定了171家医院纳入遗传亚组,在这些医院入组的12 603例患者参与了基因测序项目,其中有1308例没有提供足够的白细胞。
2.1.2 DNA提取 常用的将基因组DNA从细胞中提取出来的方法有两种,其一是采用酚氯仿法提取,其二是使用具有独特分离作用的磁珠(如DP329磁性血液基因组DNA试剂盒)。提取既可以手工进行,也可以使用全自动仪器(如核酸蛋白提取系统)进行。
2.2 测序数据生成
2.2.1 DNA质量评估 提取出的基因组DNA需要经过质量评估,DNA总量、浓度和片段长度均合格的样本才可用于基因测序。常见的对基因组DNA的浓度进行定量测量的仪器有Qubit 2.0荧光仪和Gemini XPS酶标仪;对DNA质量的评估一般在琼脂糖凝胶上进行电泳,以确保基因组DNA没有大量降解。合格样本的标准见表1。在CNSR-Ⅲ项目中,经过DNA提取和质量评估,381例患者的DNA含量不足或质量不合格。
2.2.2 测序平台选择 大规模队列研究通常采用的高通量基因测序仪器为Illumina®测序仪或Ion TorrentTM测序仪[15]。出于人类遗传资源数据安全以及成本方面的考虑,国内的队列研究也逐渐开始采用国产的华大智造DNBSEQ测序平台,如ChinaMAP计划[7]。不同测序仪所用的化学试剂、扩增方式、检测方式、读段长度等均有不同,各有利弊。CNSR-Ⅲ研究的测序项目由华大制造的BGISEQ-500型号的测序仪(以下简称BGISEQ)完成。除外没有提供足够白细胞的患者以及DNA含量不足或质量不合格的样本,有10 914例患者的外周血白细胞样本进行了基因测序文库制备[16]。
2.2.3 文库制备 “文库”指的是带有可用于测序的侧翼接头的DNA片段,不同的测序仪所需要的文库大小及接头序列不同,因此文库制备方法与所选用的测序仪有关。BGISEQ的文库制备步骤包括随机打断基因组DNA,选择一定长度范围内的DNA片段,末端修复,连接接头,扩增连接产物,分离单链并与夹板寡核苷酸生成单链环化DNA,消化线性分子,最后对连接产物进行纯化以得到最终文库(质量控制标准见表1)。在CNSR-Ⅲ项目中,11例样本经反复多次尝试均无法成功制备文库,159例样本疑似有微生物污染,最终有10 744例样本进行了基因测序。
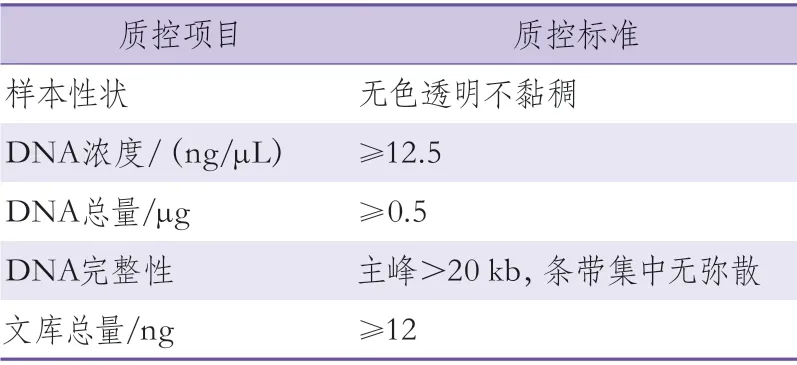
表1 基因组DNA及测序文库质控标准
2.2.4 测序 “测序”指的是应用特定技术对文库DNA进行检测并且使用与平台相关的专用软件进行初始分析及碱基检出的过程。对合格的文库进行滚环扩增以产生DNA纳米球,然后将DNA纳米球加载到规则阵列芯片上,并在BGISEQ平台上进行测序。序列衍生原始图像文件由BGISEQ基本碱基识别软件在默认参数设置下处理,序列数据以FASTQ格式存储,包含每一读段的序列以及相应的碱基质量分数。图1展示了基因测序实验部分的流程图。

图1 临床研究中基因测序项目的实验流程
2.3 测序数据生物信息学分析及质控
2.3.1 测序读段清理 “读段”指的是高通量基因测序仪检测出来的DNA片段。测序仪的原始输出文件中往往含有一些低质量的读段,需要进行过滤。如果任一读段含有测序接头,或低质量碱基比例(碱基质量≤12)超过50%,或无法识别的碱基(“N”碱基)比例>10%,则移除该读段对。之后对FASTQ文件进行质检,使用Fastp软件进一步过滤掉低质量的读段和碱基[17]。
2.3.2 基因组比对 基因测序的读段来自基因组DNA的随机打断,因此将质量合格的读段比对回人类基因组,比对情况可以反映该数据来源样本的DNA质量。利用在Sentieon软件中实现的Burrows Wheeler校准工具将读段比对到hg38人类参考基因组序列上[18],比对结果储存在SAM或BAM文件中,该文件包括读段序列、比对到染色体的位置以及比对质量等信息。完全相同的读段通常是由于文库制备时的PCR过程带来的,因此需要去除。
2.3.3 杂合度 临床研究的样本可能会存在污染,如果污染来自微生物,会体现在文库制备与测序数据质量上,在前述质控环节即应被鉴别与去除;而如果污染来自其他人源样本,则需要基于测序数据基因组比对的结果进行深入分析。使用VerifyBamID软件可以检验样本之间的污染与混杂[19]。
2.3.4 测序数据质量评估 本研究通过对CNSR-Ⅲ项目10 744例样本的数据质量进行整体评估,确定了9个单样本质控项目,这些项目的内容与阈值见表2。质量评估结果为,15例样本的10X覆盖度<80%,1例样本的错配率>0.9%,267例样本的人源污染率>0.03,这些样本均被去除。图2的上半部分展示了对每个样本进行生物信息学分析及质控的过程。

表2 中国国家卒中登记Ⅲ队列9个单样本质控项目的测序数据质控标准
2.4 遗传变异鉴定
2.4.1 单样本遗传变异鉴定 GATK(genome analysis toolkit)最佳实践指南是学术界最常用的高通量测序数据处理流程。根据该指南,首先,对样本比对基因组的结果BAM文件进行碱基质量分数重新校准,目的是使用经验误差模型调整测序读段的碱基质量分数;其次,使用Sentieon软件实现的Haplotyper算法为每个样本鉴定SNV和INDEL,最终生成VCF变异调用文件,该文件涵盖每个变异在染色体上的位置、该位置原始的序列和变异序列,以及鉴定该变异的可信程度等信息。
2.4.2 变异联合检测 变异联合检测是指同时考虑所有样本的变异检测过程,该过程能利用一些样本中的信息来推断另一些样本中最可能的基因型[2],从而提高低覆盖区域中变异检测的敏感性。利用GATK软件可以将同一项目中所有样本的变异调用文件整合为涵盖全部样本上全部变异的文件[20]。
2.4.3 变异质量分数重新校准 变异质量分数重新校准可以通过计算一个新的质量分数并以此为标准过滤遗传变异,从而平衡鉴定变异的特异性和敏感性。使用GATK软件对全部样本进行硬过滤以去除高杂合度位点[20],接着将变异分为SNV和其他变异(包含INDEL和混合变异),并分别进行变异质量分数重新校准。在CNSR-Ⅲ项目中,两类变异的过滤标准分别设定为敏感度99.0%和98.0%。图2的下半部分展示了对全部样本进行联合生物信息分析及质控的过程。

图2 临床研究中基因测序项目的生物信息分析流程
2.5 样本临床信息推断
2.5.1 性别推断 全基因组测序数据包含了个体的全部遗传信息,因此可以推断出样本的性别。常见的推断性别的原理有两种:其一为根据性染色体深度,该方法同时可以判断性染色体非整倍性;其二为根据X染色体杂合度,如可以使用Plink软件判定X染色体杂合度异常的样本[21]。
根据性染色体的深度进行性别推断的方法:对于每个样本,将X染色体和Y染色体的深度分别通过整个基因组的深度进行归一化,并表示为二维图上的一个点,两个轴的坐标分别表示归一化的X染色体深度和归一化的Y染色体深度。根据临床研究收集的基线信息对每个样本按照性别进行标记后,在二维图上自然地出现了边界。在CNSR-Ⅲ项目中,仅凭归一化的Y染色体深度0.075的简单水平线即能够将男性患者与女性患者分开,因而被选为性别推断的阈值(图3)。
同时,图3中的离群散点代表了疑似性染色体非整倍性的样本,由于纳入这一步分析的样本已经经过了测序质量的筛选,因此这些样本更有可能是人群中自然的性染色体异常患者而非测序异常导致的。为了避免异常性染色体对遗传相关机制研究产生影响,CNSR-Ⅲ项目中的这11例样本被排除。同理,Plink软件推断出的13例异常X染色体杂合度样本在后续分析中也被排除。
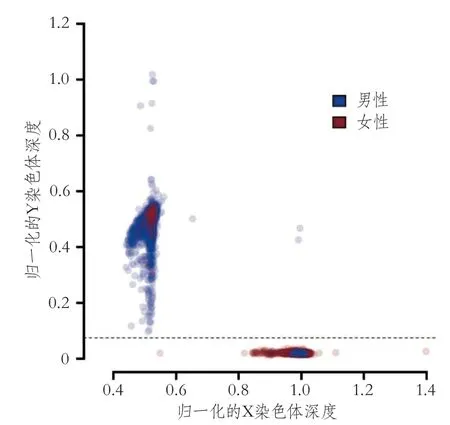
图3 根据性染色体的归一化深度对样本进行性别推断
2.5.2 亲缘关系及重复样本推断 不同的临床研究纳入的患者不同,有的是针对家系的研究,有的则认为大部分受试者均无亲缘关系,通过KING软件可以推断样本之间的亲缘关系以及群体中的重复样本对[22]。CNSR-Ⅲ队列作为一个前瞻性观察性研究,没有针对家系进行纳入或排除,因此患者之间有可能存在亲属关系。使用KING软件推断样本之间的潜在亲缘关系,对于PI_HAT>0.125的个体对认为彼此之间是遗传上相关联的。
2.5.3 临床样本管理 基因测序项目不仅要保证数据正确,还要确保测序数据与生物样本对应关系准确。因此,在纳入排除样本的流程中,需要考虑样本推断信息与临床记录不一致的情况(图4)。在CNSR-Ⅲ项目中,将通过基因测序推断的性别与临床数据库的性别进行对比,发现154例样本的推断性别与记录性别不一致,且经过中心化项目管理部门与分中心研究者的校验与核对,无法判断是数据库记录错误还是样本对应错误,因此删除这些样本。此外,经过临床研究项目管理部门的工作,所有推断出来的亲缘关系均得到了分中心研究者确认,因此将38例患者排除在队列之外。
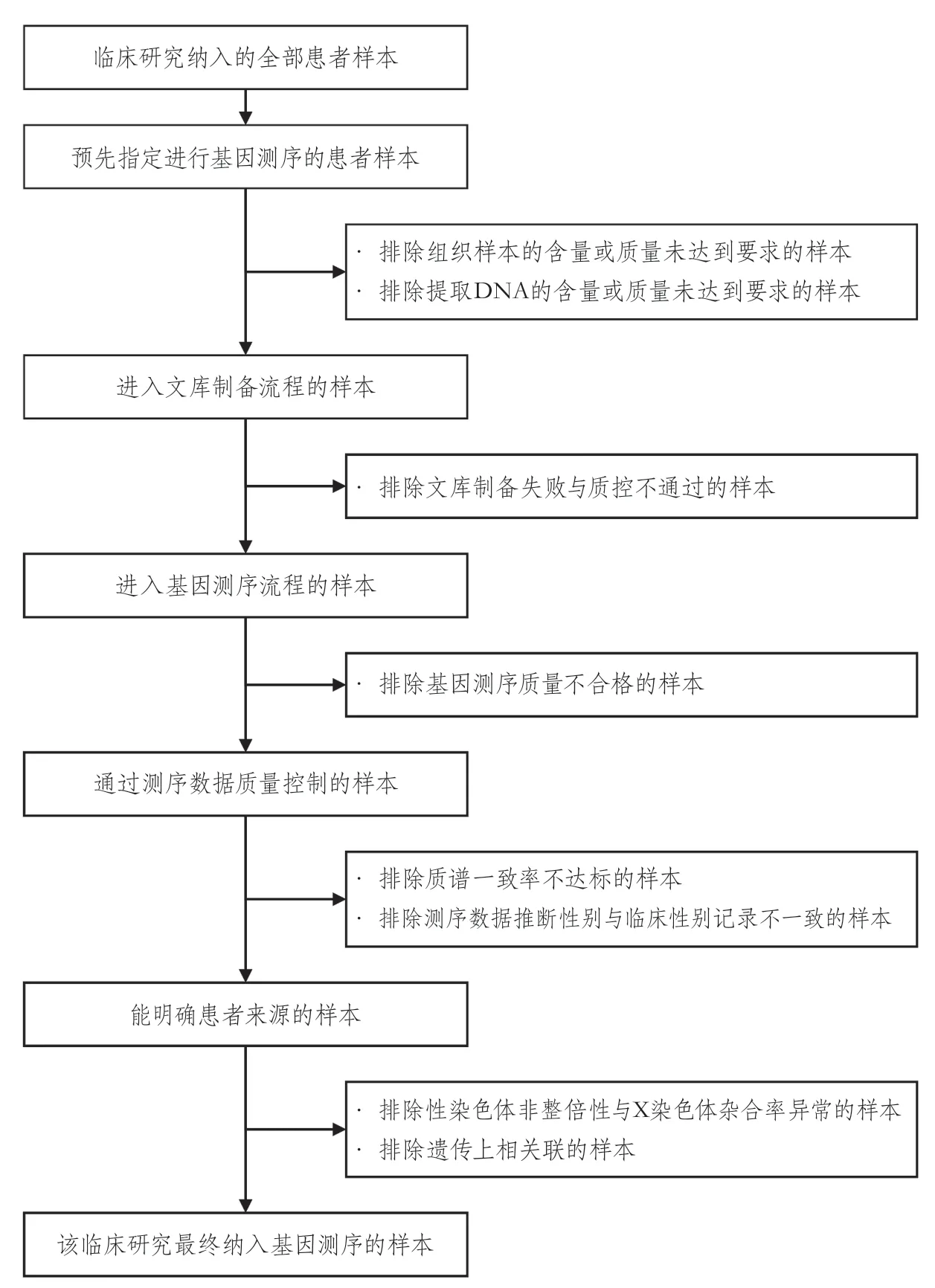
图4 临床研究中基因测序项目纳入排除样本的流程
2.6 基因测序项目的质量保证
2.6.1 基因检测数据的相互验证 对于大型临床研究的基因测序项目,为了确保样本在传递、提取、上机的过程中没有被搞混,最好安排独立平行的其他基因检测方式,以验证DNA样本和测序数据来自同一个体。在CNSR-Ⅲ项目中,选取了21个双等位基因指纹SNV,采用基于飞行质谱的基因分型方法进行项目质量管理[16],4例样本因SNV分型一致率<85%而被排除。经过上述一系列样本质控,有10 241例样本最终可用。
2.6.2 样本编号 涉及临床研究的基因测序项目,为了避免患者隐私泄露以及人类遗传资源数据安全遭到威胁,不仅需要对患者信息进行脱敏,而且建议不使用能追溯到其他信息(如患者基线、随访、生物标志物、影像等数据)的编号,可以采用一套新的唯一不重复编号并维护与其他信息编号的对应关系。在CNSR-Ⅲ项目中,以生物样本送检时的孔板编号为基础编制了基因测序编号,在DNA提取和测序、SNV分型以及生物信息学分析的整个流程中均使用该基因测序编号,在后续涉及具体课题的分析中才对应回与其他信息能够匹配的编号上。
2.6.3 样本溯源 为了便于样本溯源,应当记录每个样本在各个环节的信息。基因测序项目中需要保留的信息包括送样批次及孔板排布信息、DNA提取批次及质控信息、引物和接头名称及序列、建库批次及质控信息、测序批次及上下机时间、过程中的成功或失败记录及异常信息。如果样本有重新送样、提取、建库情况,每次重复操作时的记录也必须保留。测序过程可能会存在首次测序反应成功但数据量或质量不合格的情况,除了需记录复测、加测时的生产记录,还需记录首次测序与复测、加测批次的对应情况,便于将多次测序的结果整合分析。上述所有记录文件均须保留电子版和纸质版记录备查。
2.6.4 批次效应控制 为了尽量降低测序反应中的批次效应,整个项目过程需采用预先指定的处理程序,测序仪和操作人员尽量保持一致,反应试剂尽量为同一生产批次。如果由于客观原因无法保持一致,应尽量换用同品牌同货号的产品并记录每个样本所使用的试剂批次、批号等,方便后续在数据处理阶段去除批次效应。
2.6.5 实时反馈 临床研究的基因测序项目往往样本量大、运行时间长,如果待所有数据都接收后再进行质控,一旦发现问题,可能会给问题的解决或漏洞的弥补带来麻烦。因此,在项目运行中应当采取实时反馈的机制,每接收一批数据应立即按照指定质控流程进行处理,并将不合格样本或异常信息向检测实验室反馈,以便及时进行重测或加测,从而确保项目稳定和高效运转。
2.7 数据安全与生物样本安全
2.7.1 基因测序数据的安全 为了保障基因测序数据的安全,在生产过程中,测序仪要做到专机专用,并且项目开始前到项目结束后一定时期不可连接互联网。测序数据生成后,不可使用网络传输,可以采用硬盘等介质并进行加密传输,密码需要采用与数据不同的途径进行传递。此外,由于数据较大,为了避免传递过程中出错,在将数据拷贝到传递介质之前需要生成MD5码,将数据从传递介质拷贝到目的地后,再对MD5码进行校验,以确保数据完整和正确。基因测序成本高昂且难以再生,因此需要建立“两地三中心”的容灾备份解决方案。
2.7.2 生物样本的安全保藏 用于基因测序的生物样本及测序过程的中间产物含有人类遗传物质,需要妥善保管。因此,在项目结束后,所用到的外周血白细胞、提取好的基因组DNA和测序文库均必须返还中心化样本库,返还过程中需妥善包装、冷链运输。对于外周血白细胞,需要使用送样时的原盒原孔原排布顺序,已用完的白细胞也需要返还空管;对于基因组DNA和测序文库,需要提交孔板排布表以及内容物的体积和浓度信息。为了避免运输过程中样本管顺序被打乱,在包装之前,需对每个样本盒拍照,要求能看得清楚样本盒的编号、板孔的状态。
3 讨论
高通量测序技术的实现改变了人类对健康和疾病的认识,如癌症基因组图谱(the cancer genome atlas,TCGA)、孟德尔基因组中心(centers for Mendelian genomics)和英国UK10K项目均采用高通量测序来进行疾病机制和健康状况的研究[6,23-24]。临床研究中基因测序的样本量通常很大,检测范围更全面,因而对分析流程标准化、质控指标统一化以及项目管理精细化提出了更高的要求。本研究确定了面向临床研究的基因测序项目的设计原则,搭建了项目管理与标准化数据质控流程,并在超过万人的CNSR-Ⅲ队列进行了验证。
对质控标准的选择是临床研究基因测序项目的重点,特别是大规模多中心的临床研究,由于不同分中心的医院等级不同、设施设备不同、研究参与人员不同,因此样本质量有所参差。对于质控标准的选择,既不能太严格导致样本量缩水增加检测成本,又不能太宽松导致影响整体研究质量,需要在样本量与样本质量之间寻找到微妙平衡。本研究确定的基因组DNA质控标准适用于任何基因检测项目,数据质控标准适用于大多数高通量测序项目(“平均测序深度”和“10X覆盖率”两个项目只适用于全基因组测序)。
通过生物信息学分析鉴别出的样本性别有时与临床信息中记录的不同,亲缘关系推断出患者中隐藏的家庭和人口结构信息也不为研究者所掌握。此外,重复样本可能暗示在样本的传递、提取、上机的过程中有纰漏。上述不一致、不明确的情况均需反馈到中心化项目管理部门进行核验与纠正,有时也需要回到分中心与研究者进行确认。这些现象在大型临床研究中很难避免,也被其他研究所报道[7]。只要问题样本所占的比例在一定范围内,这其实不是坏事,通过对临床研究的项目管理以及样本质量形成闭环反馈,可以帮助找到并排除与临床信息不对应的生物样本,降低统计分析中的假阳性与假阴性。
本研究在基因测序项目的质量保证方面所做的工作(基因检测数据的相互验证、样本重编号、全流程记录、实时反馈等),尽最大可能降低了批次效应和系统偏差,保证了生物样本安全与数据安全。然而,本研究还存在以下局限性:第一,样本到达中心化生物样本库之前的步骤可控性差,院内采集患者生物样本以及运输过程中均存在弄混样本的可能;第二,项目运行中人员、试剂、仪器等的更换难以控制;第三,尽管已经进行了非常详尽的记录,仍不可能穷尽批次效应的所有潜在来源。后续通过专门考虑批次效应的统计分析方案,可以进一步降低乃至消除批次效应[10],而这也依赖于本研究产生的可溯源性记录。
总之,本研究所搭建的基因测序管理流程在CNSR-Ⅲ项目中应用成功,也为其他临床研究中基因测序项目的管理与质量控制提供了参考与借鉴。
说明:本文涉及的部分生物信息学术语或数据库名称在国内尚无统一译文,强行将这些术语或名称翻译成中文将影响读者对原意的理解,因此本文对此类术语及名称未进行翻译。
【点睛】本研究搭建了一套基因测序项目框架,在中国国家卒中登记Ⅲ队列上应用成功,可以为其他临床研究中基因测序项目的管理与质量控制提供参考与借鉴。
猜你喜欢
军事文摘(2022年16期)2022-08-24
今日农业(2022年4期)2022-06-01
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
科学导报(2021年29期)2021-06-03
支部建设(2020年15期)2020-07-08
科海故事博览·下旬刊(2019年6期)2019-04-16
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
雕塑(1996年4期)1996-07-12
