
主族元素AB4型含氧酸根的成键分析
——推荐一个研究型计算化学实验
2022-03-30 01:31许文华李安阳岳可芬
大学化学 2022年2期
许文华,李安阳,岳可芬
西北大学化学与材料科学学院,化学国家级实验教学示范中心(西北大学),西安 710127
化学键是化学中用于表示原子或分子之间吸引力的广泛符号。然而化学键并不是在严格意义上可观察到的,所以我们用化学键模型表示。化学键模型,诸如共价键、离子键、金属键、氢键、配位键等,在化合物分子的分类方面发挥过巨大作用,现在依然用于解释和指导实验[1]。价键理论和分子轨道理论是处理化学键尤其是共价键的基本理论。
对于主族元素AB4型含氧酸根价键理论认为中心原子(Si、P、S和Cl)采用sp3杂化与每个O原子成σ键,很好地解释了其正四面体的结构;再引入d轨道参与成键,d轨道在原来的σ键的基础上形成dπ-pπ键,使得原来的键增强,键长短于常规单键,与实验事实符合。这作为典型的dπ-pπ成键例子被写进教科书[2]。然而主族元素原子d轨道是否参与成键,一直有不同意见和讨论[3,4]。分子轨道理论认为除了轨道对称性的匹配,轨道能级是否接近也是判断轨道之间能否成键的基本条件之一。多数的理论研究均表明,主族元素的d轨道由于能量较高,难以参与成键[5]。若d轨道不参与成键,化学键也可以因为其他作用而强化,譬如π →σ*超共轭[6]。AB4型含氧酸根的A―B键略强于单键,是由于形成了dπ―pπ键?还是源于其他相互作用?需要通过定量的方式给予解释,基于分子轨道方法的计算是一个合适的选择。
在分子轨道理论的框架下,给出所有轨道的信息,观察其对称性和能量,原则上是可以整理清楚原子之间的关系。然而分子轨道理论缺乏化学形象,化学相关专业的学生理解起来相对困难。对于学过结构化学课程的高年级本科生,可以通过计算化学的方法做成键分析,加深对分子轨道理论以及化学键的理解。SO42-这类简单体系的理论计算不需要高级的资源,目前在个人电脑上短时间内也可以完成。近年来,国内多所高校均已开展计算化学的实验教学[7,8],西北大学也于2017年为三年级本科生专门开设计算化学实验课程。在基本操作类实验之后,研究型实验对提高学生能力、促进学生勤于思考,进而培养学生对知识的综合分析和应用能力会有很大的帮助。为此,针对AB4型含氧酸根中A―B键的异于常规单键的问题,设计计算化学实验,基于分子轨道理论,用不同的方法分析其化学成键。
1 实验目的
(1)复习并进一步理解分子轨道理论;
(2)熟练掌握图形化分子轨道的方法;
(3)学习运用分子轨道理论分析化学键;
2 实验原理
2.1 正则轨道和定域轨道[9]
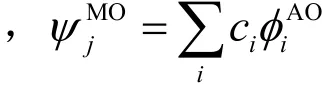
2.2 化学键分析
目前最流行的分析化学键的方法是自然键轨道(Natural Bond Orbital,NBO)分析和分子中的原子(Atoms in Molecules,AIM)分析。尽管它们并不是将CMO变换成完全意义上的LMO,但与分子轨道定域化很大程度上是相似的,很适合做化学键的初步分析。
NBO分析方法从电子结构计算得到的一阶约化密度矩阵出发,得到路易斯结构(Lewis structure)的化学成键,即内层、成键、反键、孤对电子和高能量的未占据的Rydberg轨道[10,11]。基于定域的路易斯结构的微扰分析可以通过计算占据和未占据轨道之间的相互作用得到。不同于NBO方法的基于原子轨道的Fock空间分析,AIM或者QTAIM (Quantum Theory of Atoms in Molecules)分析是对实空间的电子密度函数做拓扑分析[12]。得到的临界点等信息中可以判断化学键的类型、强弱等。类似的拓扑分析方法可以扩展到其他基于密度的函数,比如反映电子局域行为的Localized orbital locator(LOL)函数[13]。
2.3 AB4型负离子
AB4型含氧酸根的稳定构型是正四面体结构,其四个A―B键相同,但键长小于单键的原子半径的加和。例如的S―O键长为149 pm,比共价单键的原子半径之和175 pm缩短了26 pm。一种观点认为,中心S原子除了以sp3杂化轨道分别与四个O原子的一个p轨道形成σ键之外,S原子的d轨道还分别与O原子的其他两个p轨道对称性匹配,形成了dπ-pπ配键,从而增强了S―O键,缩短其键长。然而,3d轨道参与成键的效率需要根据具体分子做不同的推断,化学键的增强也可能源于离子相互作用,超共轭作用等。
3 实验步骤
3.1 优化的稳定构型
使用GView程序(version 6.0.16)搭建分子并保存为输入文件。在文本编辑器中打开并修改保存此输入文件,用密度泛函方法优化来得到气相下该离子的稳定结构,选择B3LYP/6-31G(d)的计算方法;计算振动频率以验证此结构为势能面上的局部极小点;保存计算临时文件chk。示例输入见表1。
表1 稳定构型优化的输入文件
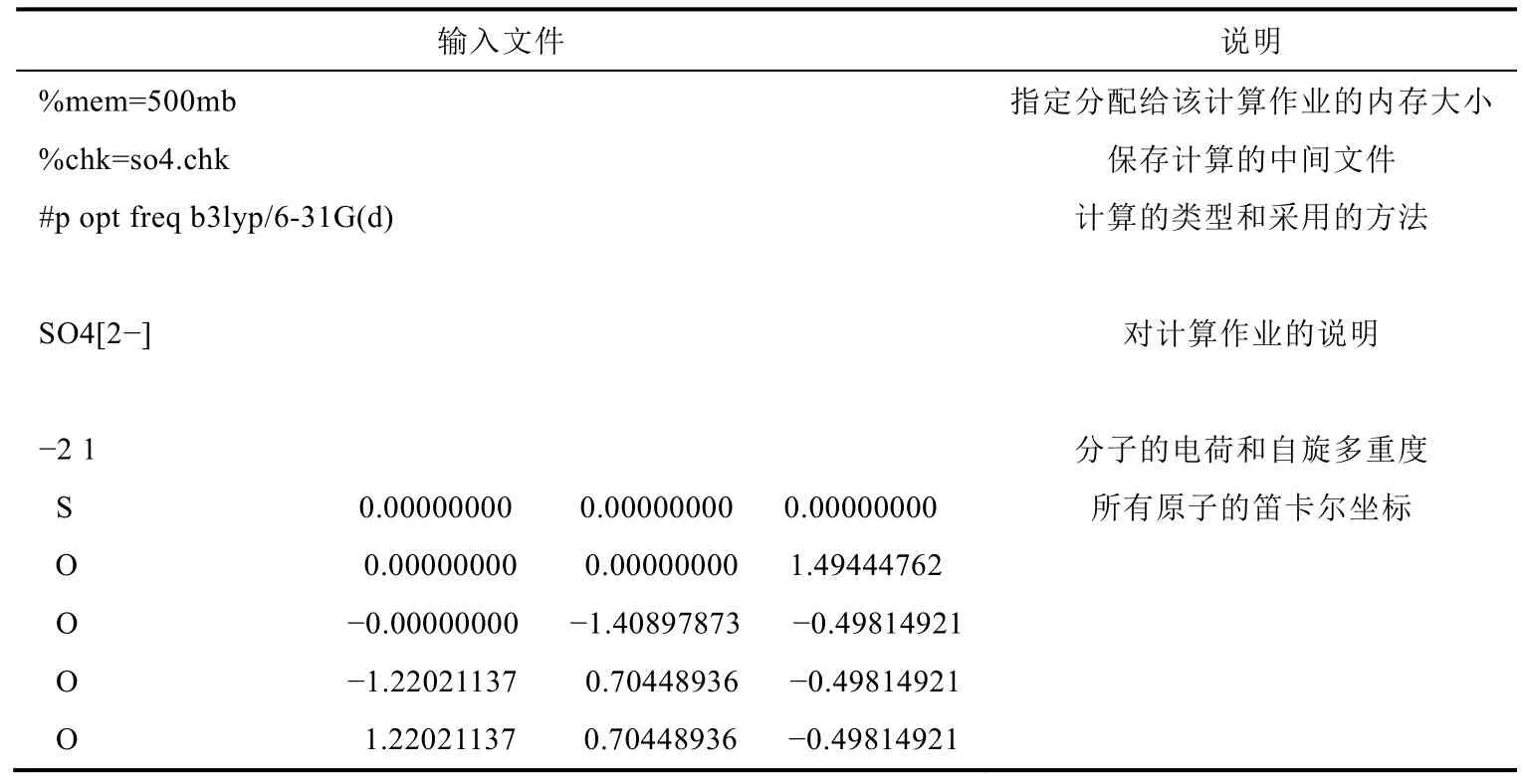
表1 稳定构型优化的输入文件
%mem=500mb输入文件说明指定分配给该计算作业的内存大小%chk=so4.chk 保存计算的中间文件#p opt freq b3lyp/6-31G(d)计算的类型和采用的方法SO4[2-]对计算作业的说明-2 1 S 0.000000000.000000000.00000000 O 0.000000000.000000001.49444762 O-0.00000000-1.40897873-0.49814921 O-1.220211370.70448936-0.49814921 O 1.220211370.70448936-0.49814921分子的电荷和自旋多重度所有原子的笛卡尔坐标
将输入文件提交到高斯程序(Gaussian version 09.E01)中运行[14]。在程序正常结束后,检查计算并分析。首先用文本编辑器或者GView程序打开输出文件(*.log或者*.out)查看振动频率,所有频率值均应为正值;查看优化好的键长,并和实验值(149 pm)对比。之后用GView程序打开chk文件,查看前线分子轨道。
3.2 NBO计算
用GView程序打开优化后的输出文件,保存为新的输入文件。修改此输入文件以完成NBO计算:增加Population关键词中与NBO有关的选项,输入文件第三行变为“#p b3lyp/6-31g(d) pop=(nboread,savenbo)”,并在分子构型输入后空一行,然后输入“$nbo bndidx $end”以计算基于轨道的Wiberg键级。
将输入文件提交到高斯程序中运行。程序正常结束后,用GView程序打开chk文件,查看成键(BD),孤对(LP)以及占据数相对较大的里德堡(RY) NBO轨道。打开输出文件,查看二阶微扰分析(Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis),找出二阶微扰作用能,重点关注能量较大的给体和受体轨道。
3.3 AIM分析
AIM分析是本实验的选作部分,为有兴趣的同学提供另一种分析化学键的方法,可以对比验证。
将3.1小节得到的chk文件转化为fchk文件并在程序Multiwfn (version 3.7)[15]中打开。
(1)选择Localized orbital locator (LOL)函数做拓扑分析。从最开始的主菜单,依次选择“2 Topology analysis”→“-11 Delete results and reselect real space function”→“10 Localized orbital locator (LOL)”。
(2)搜寻LOL函数的临界点(Critical Points, CPs)。接(1),依次选择“6 Search CPs from a batch of points within a sphere”→“-1 Start the search using each nucleus as sphere center in turn”。
(3)显示CPs。接(2),依次选择“-9 Return”→“0 Print and visualize all generated CPs, paths and interbasin surfaces”。在可视化窗口中,选择(3,-3)类型的CP点,并保存该图片。
3.4 数据分析及讨论
表2 化学成键相关数据

表2 化学成键相关数据
性质 数据 说明S―O的键长152 pm 实验参考值为149 pm S―O的键级1.09基于NAO轨道的Wiberg键级S―O σ成键轨道的组成32 (25/73/2) + 68 (24/76)百分比表示,格式:S (3s/3p/3d) + O (2s/2p)sp类型的孤对电子轨道100 (76/24)百分比表示,格式:O (2s/2p)p类型的孤对电子轨道100 (0/100)百分比表示,格式:O (2s/2p)S―O σ*反键轨道68 (25/73/2) + 32 (24/76)百分比表示,格式:S (3s/3p/3d) + O (2s/2p)基于NBO的二价微扰能52-70 kJ·mol-1O的LP-p轨道和S―O的σ*反键轨道的相互作用
由于对称性,部分分子轨道的能量相同,分布类似,图1显示了10号至29号共20个正则分子轨道的波函数图像。图示的轨道是完全离域的,与我们对化学成键的形象理解没有对应关系,正则轨道不适用于成键分析。用NBO方法对分子轨道局域化之后,20个轨道重新组合成了4个类型,能量由低到高分别有4、4、8、4个轨道能量简并(见图2)。其中10-13号是4个S―O成键轨道(bond,BD),26-29号是4个反键轨道(antibond,BD*)。成键轨道中S原子的3s轨道和3p轨道成分比例大致是1 : 3,3d轨道对键的贡献非常小,几乎可以忽略(见表2)。O原子的2s和2p轨道都有参与了成键,2p轨道的成分相对更多。结合的正四面体构型,借用杂化轨道的概念,可以认为S原子采取sp3,与4个O原子形成4个σ键;但是S原子d轨道对成键的作用可以忽略,中不存在所谓的dπ-pπ键。能量介于成键轨道和反键轨道之间的12个轨道中,S原子没有任何贡献,可以认为是非键轨道,被孤对(lone pair,LP)电子所占据。LP轨道中,14-17号轨道能量略低于18-25号轨道,O原子的2s和2p轨道都有参与,并且与BD轨道中2s和2p轨道成分比例相反。可以认为每个O原子都采取sp不等性杂化,一部分与S原子形成σ键,一部分成为LP-sp轨道。18-25号轨道则完全由O原子的2p轨道组成,与邻近的S―O成键垂直。综上,由原子轨道(AO)线性组合形成的分子轨道(MO)可以用图3来表示。
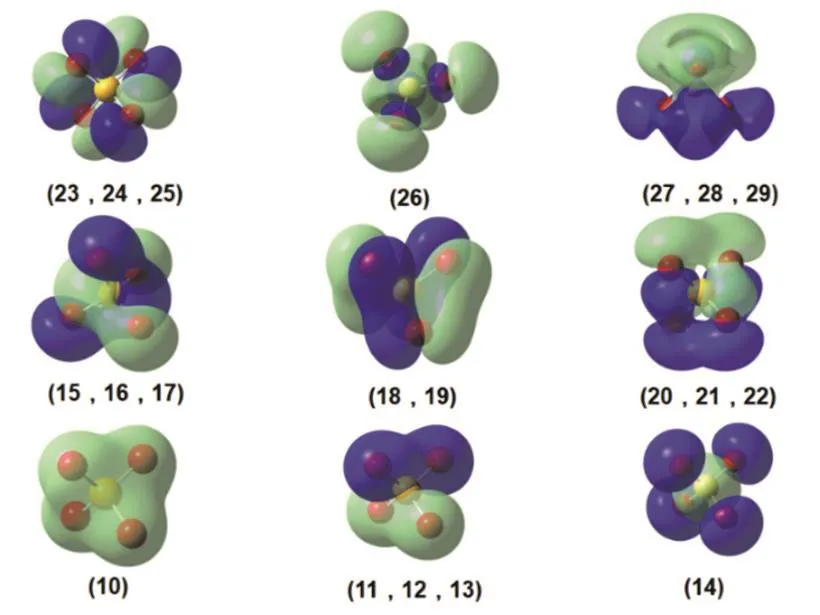
图1 的价层正则分子轨道
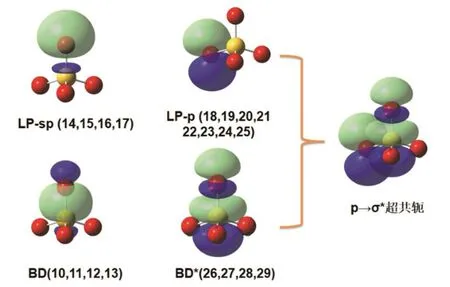
图2 的价层NBO轨道等值面= 0.03
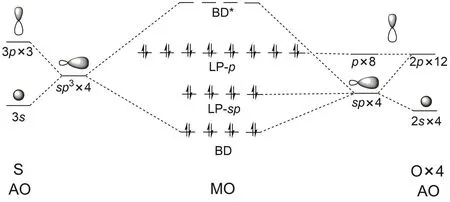
图3 原子轨道组成分子轨道(LCAO-MO)的示意图
针对密度函数做QTAIM分析,首先得到临界点(critical points,CPs)和键径(bond path)。其中临界点包括4个键临界点(bond critical points,BCPs)和5个核临界点(nuclear critical point,NCP)。在可忽略的数值误差范围内,核临界点和原子核位置重合,而键径和原子核之间的连接直线重合。这定量的还原了化学家,尤其是有机化学家们一直使用的分子的球棍模型。键临界点的键椭圆度(bond ellipticity)几乎为零,这通常意味着化学键为单键,而非双键。进一步对LOL函数做拓扑分析,结果表明中心S原子与每个O原子之间的区域存在一对成键电子,氧原子的价层另外存在三对孤对电子(见图4),验证了图3中对轨道定域化后分类的合理性。
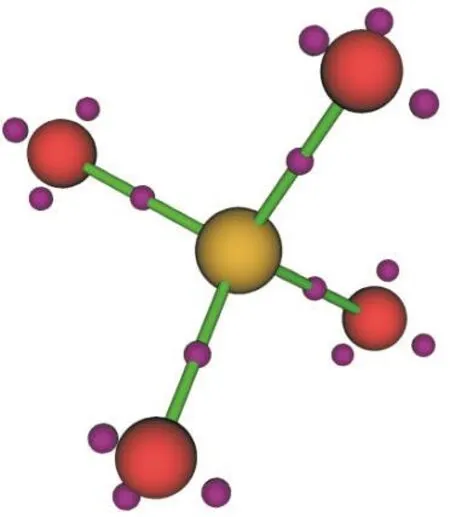
图4 的LOL函数的(3, -3)类型的CP点
然而S―O键长小于这两个原子的共价半径之和的事实并没有得到解释。Wiberg键级计算证实了中成键不是纯粹的σ单键(见表2)。既然S原子的d轨道不参与成键,对成键的贡献很可能来自于占据轨道和其他空轨道之间的超共轭作用。所谓超共轭作用可以理解为:占据的NBO轨道向相邻的非占据NBO轨道发生电子离域,令体系能量的降低。在NBO计算中,通过二阶微扰理论可以近似估计这个能量。若能量较大,可以认为两个NBO轨道之间存在超共轭作用,从而增强相应原子之间的成键。O原子的每个LP-p轨道上电子基本满占据,与相邻3个S―O的σ*空轨道的二价微扰能分别为52、53和70 kJ·mol-1,而其他类型轨道之间的微扰能最大值不足以上值的一半,它们之间存在较强的相互作用。此外,从图2可以看出,O原子的LP-p轨道与邻近的BD*轨道对称性是匹配的,而能量上,这两个轨道也是最接近的占据和未占轨道(如图3所示)。由此可以推断,p→σ*超共轭增强了S原子与O原子之间的相互作用,从而使S―O键长缩短。
3.5 其他AB4型离子的计算和分析讨论
4 实验组织和建议
计算化学实验不是纯粹的实验操作课,理论教学与实验教学同时进行。每个实验开始前,用大约1个学时的时间结合计算软件的输入文件复习相关的结构化学知识。首先学生可以明确计算的目标,用学过的知识预期实验结果,一旦实验中出现错误,可以及时自我识别和纠正;其次,实验计算出来的结果可以用来检验所学理论知识的适用范围。
在进行这个实验之前,按课程的进度,学生已经完成了5次计算量子化学初级验证性实验,较为熟悉Gaussian 09和GView软件的运用。本次实验是计算化学实验的提高部分,侧重数据的分析,因此安排学生以小组为单位完成实验,每组2-3人,协同完成计算、分析和总结报告。报告中需包含:(1)对实验体系的调研,AB4型含氧酸根化学键的特别之处,传统的解释,不足之处;(2)对比不同计算任务产生的分子轨道,讨论其相同和不同之处;(3)成键分析,对此类化学键从分子轨道的角度进行解释,NBO分析必做,AIM计算可选做;(4)小结实验过程的认识和体会。
此外,设置思考题,让学生查找资料,培养严谨科学的思维习惯。(1) NBO分析是否适用于更复杂的分子结构?譬如分子中存在大Π键。(2)这个体系的中性分子会不会有不同?原因是什么?(3)常说的HOMO、LUMO之类的轨道是CMO还是LMO?
5 结语
与实验表征方法类似,计算化学已经逐渐成为化学教学和研究的一种常用方式。本实验的开设,出发点之一就是针对结构化学教学中的疑难问题,用现代计算的手段提供一个合理的解释。从两年来在西北大学计算化学实验课上的实践来看,学生能够一步步地完成计算要求,加深对分子轨道基本概念的理解,进而用于分析化学成键作用。对计算化学这一新技能的掌握也因为实验的练习进一步提高。需要指出的是,只要是格式正确的输入,计算机都会给出一定结果,但计算出来的结果是否合理可靠,则需要理论知识进行判别。本实验的选题和设计,相对来说都是比较有难度的,不仅从计算结果理解理论知识,也促使学生主动思考,学会用理论知识对计算结果深入分析。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
空间科学学报(2020年6期)2020-07-21
空间科学学报(2020年6期)2020-01-08
环球时报(2019-12-05)2019-12-05
考试周刊(2016年60期)2016-08-23
考试周刊(2016年48期)2016-06-29
中学生数理化·高二版(2016年6期)2016-05-14
太空探索(2014年4期)2014-07-19
