
过渡金属催化的6-苯基-5,6-二氢-2H-吡喃-2-酮的合成
——推荐一个大学有机化学综合实验
2022-03-30 01:31刘治彤赵子逸彭渤曲玥名田松麟俞寿云
大学化学 2022年2期
刘治彤,赵子逸,彭渤,曲玥名,田松麟,俞寿云
南京大学化学化工学院,南京 210023
1 引言
碳-碳键的形成和断裂是有机合成的中心问题。科学家们通过不断的探索研究,已经做出了许多伟大的成就。本次实验以碳-碳键连接为中心议题展开,通过两种不同的碳-碳键连接方式进行三步合成反应。可以在实验过程中体会碳-碳键形成在有机合成中的重要地位,并且了解经典的和相对前沿的碳-碳键形成的多种方式。
金属介导或者催化的反应是形成碳-碳键的强有力方法,该类反应备受瞩目。在这个领域曾有过三次诺贝尔奖级的成果,分别是1912年的“格氏反应”、2005年的“烯烃复分解反应”以及2010年的“钯催化的交叉偶联反应”。格氏反应已经进入本科生理论和实验的学习,但在实验过程中,存在格氏试剂较难引发、引发现象不很明显等一系列困难,因而,本实验提出一种改进的方式,更好地完善格氏反应的实验教学。目前已有部分国内外高校在本科化学实验中开设了钯催化的交叉偶联反应的实验教学内容[1]。迄今为止,烯烃复分解反应在本科理论教学中少有涉及,实验教学中更是缺少这一诺奖级的成果。因而,本实验设计了一种适用于本科教学的烯烃复分解反应,有利于学生更好地全面了解有机化学的重要发展历程和成果。
最为经典的碳-碳键的形成反应当属格氏反应。1912年,诺贝尔化学奖授予了Victor Grignard。Grignard借鉴了Frankland和Wanklyn制备有机锌试剂的方法,在室温、常压下成功完成了镁试剂的制备并和酮反应制备醇,该成果于1900年发表[2]。格氏反应已经较为成熟,在基础有机化学实验中多有涉及。但是基础实验教学中经典的格氏试剂引发过程不是很容易,且不易判断。本实验尝试改进,通过反应溶液颜色的明显变化指示反应的进程。
烯烃复分解反应是前沿的碳-碳键形成反应,是连接sp2-碳和sp2-碳的高效方法。从反应的净结果来看,是两个烯烃在催化剂的作用下相互交换,因而也被形象地誉为“交换舞伴的反应”。烯烃复分解反应是2005年诺贝尔化学奖的成果。最初,该反应在工业界被发现[3]。如今,烯烃复分解反应,特别是关环复分解反应(Ring Closing Metathesis,RCM),为众多天然产物的合成提供了便捷的路径[4]。学习并深入了解该反应,可以拓宽有机合成的思路。最初烯烃复分解反应被发现时,虽然在有机合成以及工业界有巨大的应用潜力,但是催化剂体系对于水和氧气不稳定,限制了其推广。Richard R.Schrock教授和Robert H. Grubbs教授研究并且发明了更加高效稳定的催化剂,为烯烃复分解反应的应用做出了巨大贡献。
Schrock教授于1980年发现了钽卡宾配合物具有烯烃复分解的能力[5]。随后Schrock教授所研发的钼或钨的催化剂有更好的反应活性[6],其中图1A中的催化剂已经商品化。Schrock教授的工作使得烯烃复分解反应的推广成为可能,为后续新一代催化剂的出现打下了坚实的基础。虽然钼或钨的催化剂活性较好,但是,对于空气和水仍然很不稳定。1992年,Grubbs教授发现钌卡宾金属化合物能够进行降冰片的开环聚合反应,且该催化剂能够对水、空气等稳定[7]。钌催化剂的出现为烯烃复分解反应打通了另外一条道路。该催化剂见图1B。1996年,Grubbs对钌催化剂进行改进,成为了第一代Grubbs催化剂,其催化性能相较于之前的催化剂更好,其结构式见图1C[8]。随着研究的深入,1999年,Grubbs发现催化剂的活性与其膦配体的解离有关,认为催化循环过程中经过一个高活性的单膦中间体,从而根据这样的事实发明了第二代催化剂,其结构式见图1D[9]。催化剂用量更少,适用于开环复分解以及关环复分解反应。
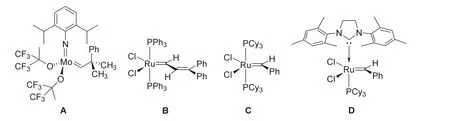
图1 常用的烯烃复分解催化剂
本实验以6-苯基-5,6-二氢-2H-吡喃-2-酮(6)作为目标化合物。使用Cp2TiCl2催化的烯丙基溴(1)对苯甲醛(2)的加成反应,4-二甲氨基吡啶(DMAP)催化的醇(3)与丙烯酰氯(4)的酯化反应和Grubbs II催化剂催化的烯烃(5)的关环复分解反应作为关键反应,合成吡喃酮(6)。实验步骤如图2所示。

图2 6-苯基-5,6-二氢-2H-吡喃-2-酮的合成的反应步骤
2 实验目的
(1)通过查阅文献,了解过渡金属催化有机反应的历史和现状。
(2)熟悉并了解Cp2TiCl2催化的醛的加成反应,DMAP催化的酯化反应和烯烃复分解反应的原理。
(3)掌握简单无水无氧的操作。
(4)掌握利用TLC监测有机反应和利用快速柱层析纯化反应产品。
(5)学会通过核磁共振谱图鉴定反应产物。
(6)巩固减压过滤、萃取、分液等基本有机实验操作。
(7)掌握旋蒸仪的使用方法。
3 试剂和仪器
3.1 反应试剂
二氯二茂钛,4-二甲氨基吡啶(DMAP),Grubbs II催化剂(其结构式见图1D)。以上试剂采购自毕得医药。烯丙酰氯,无水四氢呋喃(有分子筛),二氯甲烷(分析纯),乙酸乙酯(EA,分析纯),石油醚(PE,分析纯)。以上试剂采购自安耐吉。其余试剂还包括:烯丙基溴(国药试剂),苯甲醛(阿达玛斯试剂),活化锌粉(自制),三乙胺(上海泰坦化学有限公司),高纯氮气(南京宁卫医用氧气有限公司),无水二氯甲烷(有分子筛,百灵威),饱和氯化铵溶液(其中,氯化铵采购自西陇化工股份有限公司),饱和食盐水(其中,氯化钠采购自西陇科学股份有限公司),乙醚(分析纯,南京化学试剂股份有限公司),无水硫酸镁(永华科学科技有限公司),磷钼酸溶液(配制方法:10 g磷钼酸+ 100 mL乙醇)。
3.2 实验仪器
分析天平,磁力搅拌器,紫外灯(254 nm),旋转蒸发仪,400 MHz核磁共振仪(CDCl3为氘代试剂,TMS为内标物)。仪器的型号、厂家和国家见表1。

表1 实验仪器汇总
4 实验步骤
4.1 Cp2TiCl2催化的烯丙基溴对苯甲醛的加成反应[10]
准备一个100 mL梨形瓶,在其磨口上用翻口橡胶塞塞紧并用密封胶带在橡胶塞与瓶口的接触部位缠绕数圈。用双排管抽换气3次并充入氮气。准备好两个氮气球。将上述准备好的气球插在置换好气体的梨形瓶橡胶塞上,用1 mL注射器在上述梨形瓶中加入苯甲醛(6 mmol,0.64 g),烯丙基溴(15 mmol,1.81 g)并用注射器加入30 mL无水四氢呋喃,配制成待用溶液。
在另一个100 mL梨形瓶中放入适当大小磁子,加入二氯二茂钛(0.06 mmol,14.9 mg)和活化锌粉(15 mmol,0.98 g)。磨口处塞上翻口橡胶塞并用密封胶带缠绕数圈,通过针头和双排管进行抽气以及氮气置换,此步骤重复三次后,用氮气保护体系,并插上氮气球。室温下开启磁力搅拌,并用注射器加入无水四氢呋喃30 mL(反应装置见图S1)。溶液颜色成为红色,等待体系颜色变绿后(大概需要10 min左右) (第一步反应颜色变化见图S2),注射入之前配制好的苯甲醛和烯丙基溴混合溶液。反应持续20 min。用TLC监测反应是否完成(原料点使用苯甲醛;展开剂可以用V(PE) :V(EA) = 9 : 1)。若未反应完成,继续反应10 min,再次使用TLC监测反应是否完成。在实验报告本上按原比例大小绘制TLC分析结果并分别计算Rf值(该反应的TLC分析结果见图S3)。反应的化学方程式如图3所示。

图3 Cp2TiCl2催化的烯丙基溴对苯甲醛的加成反应
反应结束后,拔去橡胶塞,缓慢滴加饱和氯化铵溶液20 mL淬灭反应。减压过滤除去沉淀,得到的溶液用20 mL乙醚分层,在分液漏斗中分离水相和有机相,水相再用乙醚萃取三次,每次用10 mL,合并有机相。有机相再用饱和食盐水洗三次,每次用10 mL。洗过后的有机相用无水硫酸镁干燥。过滤去除固体沉淀,溶液接在一个250 mL的茄形瓶中。调整旋转蒸发仪的水浴温度为35 °C左右,将茄形瓶装在旋转蒸发仪上并用塑料夹固定。开启泵后调节压力并旋转旋钮使体系密封,先缓慢开启旋转,待溶剂开始蒸出且没有爆沸后,将茄形瓶放入水浴中加速旋转,快速蒸除溶剂。
使用快速柱层析纯化产物。淋洗液使用V(PE) :V(EA)从50 : 1到20 : 1到10 : 1梯度洗脱。产物为无色油状液体3。称重,计算产率(0.78 g,产率88%)。用NMR鉴定,谱图见补充材料图S4和S5。1H NMR (400 MHz, CDCl3):δ7.35-7.25 (m, 5H),5.86-5.75 (m, 1H),5.19-5.11 (m, 2H),4.74-4.71(dd,J= 4.8, 8.0 Hz, 1H),2.56-2.45 (m, 2H)。13C NMR (101 MHz, CDCl3):δ143.93,134.51,128.40(2C),127.56,125.86 (2C),118.38,73.35,43.83。
4.2 DMAP催化的醇与酰氯的酯化反应(图4) [11]
在25 mL两颈瓶中加入适当大小磁子,加入DMAP (0.2 mmol,24.4 mg),在两颈瓶上磨口处连接滴液漏斗,将滴液漏斗上方和两颈瓶另一瓶口用翻口橡胶塞塞住并用密封胶带缠绕数圈,进行氮气置换并用氮气保护体系。橡胶塞上插入氮气球后,室温搅拌下依次从两颈瓶侧瓶口处注射加入5 mL无水二氯甲烷,第一步产物3 (4 mmol,0.59 g),三乙胺(8.8 mmol,0.89 g)。在滴液漏斗中注射酰氯4 (8 mmol,0.72 g),冰浴下小心缓慢滴加酰氯入反应体系,在20-30 min内滴加完成(反应装置见图S6)。撤去冰浴,室温下反应3 h,用TLC分析反应是否完成(该反应的TLC分析结果见图S7)。(展开剂可以用V(PE) :V(EA) = 9 : 1)如果没有完成可以继续反应20 min后监测。反应的化学方程式如图4所示。

图4 DMAP催化的醇与酰氯的酯化反应
反应结束后,拆除滴液漏斗等装置,缓慢滴加10 mL饱和食盐水淬灭反应,转移至分液漏斗中并用5 mL二氯甲烷分层,水相用二氯甲烷萃取三次,每次5 mL。合并有机相,并用饱和食盐水洗涤三次,每次5 mL。有机相用无水硫酸镁干燥,过滤固体后,溶液用旋转蒸发仪除去溶剂。使用快速柱层析纯化产物。淋洗液使用V(PE) :V(EA)从50 : 1到20 : 1到10 : 1梯度洗脱。产物为无色液体5。称量,计算产率(0.58 g,产率69%)。用NMR鉴定,谱图见补充材料图S8、S9。1H NMR (400 MHz, CDCl3):δ7.35-7.26 (m, 5H),6.45-6.40 (dd,J= 1.6, 16.8 Hz, 1H),6.19-6.11 (dd,J= 10.0, 17.2 Hz, 1H),5.90-5.86 (dd,J= 5.6, 7.6 Hz, 1H),5.84-5.81 (dd,J= 1.6, 10.4 Hz, 1H),5.77-5.66 (m, 1H),5.11-5.03(m,2H),2.74-2.56 (m, 2H)。13C NMR (101 MHz, CDCl3):δ165.33,139.98,133.21,130.85,128.60 (2C),128.44,127.98,126.53 (2C),118.11,75.34,40.76。
4.3 Grubbs II催化剂催化的烯烃关环复分解反应(图5) [12]
在150 mL两颈瓶中加入适当大小磁子和Grubbs II (0.056 mmol,47.5 mg,结构式见图1D),两颈瓶上磨口处连接上球形冷凝回流管后,所有接口处塞上翻口橡胶塞并用密封胶带密封。体系置换氮气并用氮气球保护。在两颈瓶侧瓶口处加入无水二氯甲烷60 mL,加入第二步反应产物(0.56 mmol,0.11 g) (反应装置见图S10)。在45 °C下搅拌4 h。用TLC分析(展开剂可用V(PE) :V(EA) = 5 : 1) (该反应的TLC分析结果见图S11)。反应的化学方程式如图5所示。
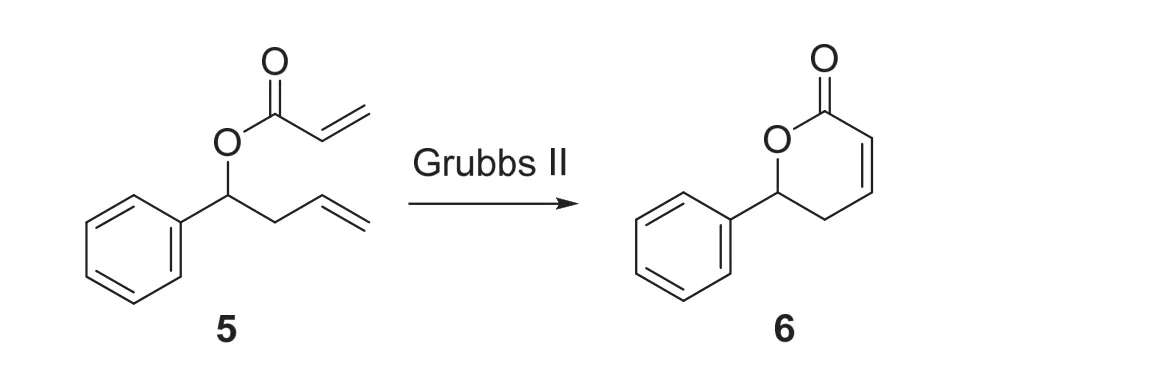
图5 Grubbs II催化剂催化的烯烃关环复分解反应
用10 mL饱和食盐水淬灭反应,10 mL二氯甲烷分层,水相用二氯甲烷萃取三次,每次5 mL,有机相用饱和食盐水洗涤三次,每次5 mL。有机相用无水硫酸镁干燥,过滤固体后旋蒸溶剂,使用快速柱层析纯化产物。淋洗液用V(PE) :V(EA)从5 : 1到3 : 1梯度洗脱。产物为白色固体6 (0.07 g,产率73%)。称重,计算产率。并用NMR鉴定,谱图见补充材料图S12、S13。1H NMR (400 MHz, CDCl3):δ7.43-7.33 (m, 5H),6.99-6.94 (ddd,J= 3.2, 5.6, 9.6 Hz, 1H),6.16-6.12 (ddd,J= 1.2, 2.0, 9.2 Hz, 1H),5.48-5.43 (dd,J= 5.2, 11.2 Hz, 1H),2.72-2.57 (m, 2H).13C NMR (101 MHz, CDCl3):δ164.01,144.82,138.47,128.69 (2C),128.64,126.05 (2C),121.77,79.25,31.60。
4.4 实验注意事项
(1)实验中涉及用双排管的抽换气过程,双排管装置图、制作氮气球、移取无水溶剂的操作具体细节见补充材料图S14-S16。
(2)本综合实验反应均在无水无氧的体系下进行,因此在用注射器加入液体的过程中,尽量减少将空气注射入反应体系,导致对实验结果造成较大的影响。
(3)薄层层析技术以及柱层析技术具体细节见补充材料中操作指南细节解说。
(4)酰氯对水和空气很不稳定,须在通风橱内操作,尽量减少其暴露在空气中的时间,并且需保证反应仪器充分干燥,不能有水残留,在称量酰氯时可以先在通风橱内加在小号塑料离心管中,封好塑料离心管盖子后去称量台称量。
(5)第二步反应体系溶液分层不易,在萃取洗涤过程中会出现乳化现象,等待时间较长,必要时可以进行离心操作;该步反应有机相在下层。
(6)柱层析中梯度洗脱的方法为:先用小极性的淋洗液冲洗过柱至杂质点先被洗脱出来,之后换用极性更大的淋洗液进行淋洗,使得所需要的产物快速从硅胶柱中洗脱下来。
5 结果和讨论
5.1 Cp2TiCl2催化的烯丙基溴对苯甲醛的加成反应
格氏反应是高效形成碳-碳键的方法之一,但是一般传统的格氏反应引发较为困难,对于不同的底物,引发所需要的温度条件等不尽相同,且是否引发成功较难判断,反应的总时间也相对较长。在反应引发阶段常会加入碘与卤代烃的卤素原子进行交换,便于引发。2012年,Brandon L.Ashfeld教授尝试选择一种催化剂,与卤代烃发生氧化加成,从而制备出金属有机化合物,进行后续碳-碳键的形成[10]。具体机理见图6。在催化循环的过程中,首先是金属(本实验中为锌)将二氯二茂钛中的四价钛还原至三价,从而三价钛可以还原卤代烃,断裂C―X键,生成C―Ti键。接着,Zn再次将四价钛还原至三价,以便可以和二价锌发生转金属过程得到有机锌试剂,最后完成与羰基化合物的亲核进攻得到终产物。而三价钛则进入下一次循环。

图6 二氯二茂钛催化循环的可能机理
该方法的实验现象非常明显,加入二氯二茂钛,使得反应体系的颜色成红色,当引发完成后,体系成为绿色,通过目测即可判断引发是否完成。总共只需要半小时就可以完成90%左右的转化。催化剂二氯二茂钛的用量仅为1%-2% (摩尔分数),且较易储存,不易变质,价格也不是很昂贵,适合本科实验教学。
重复实验的过程中发现该反应引发的时间与反应物的量有一定的关系,随着反应物增多,引发所需要的时间相应延长。但由于实验现象明显,即发生颜色的改变,可以根据这一实验现象进行判断,适当增多或减少原料的用量。
温度和季节的变化对于该反应也有一定的影响。在空气湿度大的季节重复实验,所得到的结果产率会稍微偏低,在做实验的过程中就要注意抽换气的次数可以更多一些,尽量让反应体系中少一些空气以及水蒸气。随着温度的升高,引发过程逐渐变快,在刚加入溶剂,即无水四氢呋喃的时候就可以看见溶液呈现较浅的绿色。为了使反应完全,引发过程至少不少于5 min。
该步反应进行多次重复,收率范围在84%-92%之间。
5.2 DMAP催化的醇与酰氯的酯化反应[11]
最常见的酯化反应通常是由羧酸和醇在强酸催化下进行反应,但该反应为可逆反应,难以转化完全。需要通过其他途径,如不断移除产物或加入过量酸、醇等方法提高产率。除此以外,还可以通过酰氯或酸酐与醇进行反应,由于酰氯和酸酐的反应活性大于羧酸,用酰氯或酸酐反应,可以更好地提高转化率,得到更多预期产物。本实验采用酰氯和醇进行第二步酯化反应,以期更快完成,产率也更高。
该反应使用DMAP作为催化剂。DMAP上有两个氮原子,其中三级胺上的氮原子可以将自己的孤对电子共振到吡啶环上,而吡啶环上氮原子的孤对电子相对定域,使得吡啶环上氮原子的亲核性增强。DMAP吡啶环上的氮原子比醇羟基上氧原子的亲核性更强,DMAP作为催化剂的催化机理如图7所示:DMAP作为亲核试剂进攻酰氯而生成酰基吡啶阳离子,醇进攻酰基吡啶阳离子的羰基得到产物酯,并同时得到质子化的DMAP。经过NEt3碱处理,再生的DMAP进入下一次的催化循环[13]。

图7 DMAP催化酯化反应机理
在该步反应过程中,由于体系成碱性,生成的酯可能在该环境下不稳定,因而会对最终的收率有一定的影响。如果反应完成后长时间不处理,可能造成一部分产物的水解,又生成醇,使产率下降,同时也对柱层析分离造成一定麻烦,因而该反应完成后需要迅速淬灭。该步反应使用高活性的酰氯,如果实验过程中对于恒压滴液漏斗的控制不当造成酰氯过快加入到反应体系中,会导致放热过快而不利于反应。根据这一特点,加入酰氯时也可以选择用注射器进行人工控制滴加酰氯,使反应体系更加稳定。
该步反应对水十分敏感,应尽量保证体系干燥,可以通过多次抽换气达到这一目的。原料醇也需要保持干燥无水,如果在空气中存放时间过久,可以用少量乙酸乙酯溶解重新旋蒸并抽干再进行反应。如果实验室提供的三乙胺为分析纯的试剂,可以直接使用,也可以采购无水的三乙胺来完成实验。为减小水对于反应的影响,可以多加入一点酰氯,但结果会稍有偏差。
该步反应进行多次重复,收率范围在64%-70%之间。
5.3 Grubbs II催化的烯烃关环复分解反应[12]
Yves Chauvin教授和他的学生在1971年提出了烯烃复分解的反应机理,该机理被许多实验结果所证实,如今广被有机化学家所接受[14]。Chauvin教授也因此成为2005年诺贝尔化学奖得主之一。具体反应机理见图8。反应首先是由催化剂中的金属(本实验中为钌)-碳双键和原料中的其中一根碳碳双键进行[2 + 2]环加成,生成四元环中间过渡态。接着经历一个开环过程,得到钌卡宾物种。该中间体再次和原料中另一根碳碳双键发生[2 + 2]环加成,经历四元环过渡态后,开环得到目标产物。在本实验中使用的是Grubbs第二代催化剂。

图8 烯烃复分解反应机理
在第三步反应的摸索过程中,发现反应溶剂和浓度对该反应的影响很大,具体结果如表2所示。使用二氯甲烷作为溶剂,反应完成需要4 h左右。作为教学实验,不特意强调产率的情况下,可以缩短反应时间。TLC分析结果干净,只有产物点。终产物在二氯甲烷以及乙酸乙酯中溶解度较好,旋蒸溶剂时不易出现固体,可用石油醚重结晶来获得产物进行后续分析实验。

表2 烯烃关环复分解反应的条件优化
该步反应进行多次重复,收率范围在73%-82%之间。
6 教学组织运行方式
本实验在理论上涉及新的知识较多,需要学生在实验前充分查资料了解实验的一些重要反应,如DMAP为催化剂的酯化反应、烯烃复分解反应等。只有在实验前充分理解这些原理,在实验过程中才会有更多收获。同时,也可以专门在实验正式开始前设置一节理论课,梳理一下实验中的重要知识点,开拓一下学习有机化学的思维。
实验中涉及的新操作有通过双排管进行无水无氧处理、用注射器移取液体样品、无水液体样品的注射、旋转蒸发仪的使用等等,这些实验操作在原来本科教学实验中较少出现,但在科研过程中非常有用,学习这些有用的技能是很必要的,因此也需要花一定的精力完成这类操作的教学,可以通过录制视频让学生提前观看后学习或者在实验过程中教师示范等方式进行。
本综合实验中后两步的反应时间过长,在实际的教学中可以由教师先录制好实验操作重点的视频,学生提前观看预习,并且在实验当天由教师提前称量好药品,学生到实验室直接搭建反应装置开始反应,在反应的过程中完成实验原理的讲解。也可以适当缩短反应时间,但反应收率会相应降低。
最后一步RCM反应在本次综合实验中操作难度较大,容错率较低,稍有疏忽就会使得产率下降。在实验中需要着重关注水对于反应体系的影响,无水条件越好,产率会相应越高。可以将最后一步反应设定为本次综合实验的考核反应,检验学生在“模拟”科研实验室条件下的实验水平。该步反应的产物为固体,在最终柱层析的时候可以考虑使用干法装柱、干法上样,会缩短一定的实验时间,同时也多方面训练了学生的实验技巧。
完成第一步实验需要约4-6 h,完成第二步实验需要约7-8 h,完成第三步实验需要约7-8 h。该合成的每一个反应都可独立成为一个基础有机化学实验,三步的连续反应可以作为一个综合多步合成实验。
7 创新性/特点/特色声明
(1)可视化金属有机试剂的制备;
(2)烯烃复分解反应的引入;
(3)有机化学相关的模拟科研实验训练。
8 结语
本实验以有机合成中重要的碳-碳键形成为主要研究对象进行展开。第一步反应改进了格氏反应引发困难以及不易判断引发成功与否的问题,使用Cp2TiCl2作为催化剂,更快更容易地形成有机锌试剂,并可目测判断,以较高的收率得到产物。其次,通过酯化反应,为第三步烯烃复分解反应做好准备。烯烃复分解反应作为诺贝尔化学奖级的成果,如果可以进入本科理论教学乃至实验教学的课程中,有利于学生对于最新化学进展的认知与了解。整个综合实验中不仅包含有机合成的常规训练,还增添了许多贴近科研的实验操作。可以训练学生的实验技能和动手能力,有助于学生对有机化学的认识和理解。
补充材料:可通过链接http://www.dxhx.pku.edu.cn免费下载。
猜你喜欢
化工时刊(2022年6期)2022-09-01
城市道桥与防洪(2022年3期)2022-05-08
安全与环境工程(2021年2期)2021-04-02
石油炼制与化工(2021年3期)2021-03-23
煤炭加工与综合利用(2020年6期)2020-07-17
化工时刊(2020年11期)2020-01-12
中国特种设备安全(2019年9期)2019-12-03
分析化学(2019年3期)2019-03-30
科技与创新(2016年5期)2016-03-17
山东工业技术(2015年6期)2015-07-27
