
谷氨酸棒杆菌生产异亮氨酸辅因子策略及其基因组整合研究进展
2022-03-30 06:50:22靳鑫王苏蒙祁庆生梁泉峰
生物技术进展 2022年2期
靳鑫,王苏蒙,祁庆生,梁泉峰
山东大学微生物技术国家重点实验室,山东 青岛 266237
L-异亮氨酸是人体8 种必需氨基酸之一,属于三大支链氨基酸。在人体生命活动中具有重要作用,因此广泛用于食品、药品、保健品、化妆品等领域[1-4]。目前生产L-异亮氨酸方法主要包括蛋白提取、化学合成、酶合成和微生物发酵[5-6]。其中微生物发酵法是工业生产L-异亮氨酸的主流方法,生产菌株主要包括大肠杆菌、谷氨酸棒杆菌、黄色短杆菌和乳酸发酵短杆菌。相比大肠杆菌,谷氨酸棒杆菌属于食品安全级微生物(generally recognized as safe,GRAS)[7-8],产物只有一种同分异构体,且不含有内毒素,具有非致病性,安全性较高,表明谷氨酸棒杆菌是生产食品药品级发酵产品的理想菌株[9]。谷氨酸棒杆菌作为L-异亮氨酸生产菌株,在氨基酸后期提取等方面优势明显。
L-异亮氨酸通过微生物生产过程中,由于L-异亮氨酸较长的代谢合成途径以及复杂的代谢调控网络,使得通过理性的代谢工程手段改造模式菌株,构建L-异亮氨酸优势生产菌较为困难。因此,主流构建高产菌的策略是通过随机诱变野生型菌株,然后筛选具有高产L-异亮氨酸潜力的诱变菌株[10],再通过理性的代谢工程策略与手段进一步改造,获得能够应用于工业化生产的高经济效益和高产量的L-异亮氨酸生产菌株[11]。但传统诱变育种存在筛选通量大、周期长和后期提取复杂等问题,并且经过传统诱变后的菌株基因组上很可能引入了有害突变,造成生产菌株遗传背景不清楚[12],导致菌株在进行下一阶段代谢工程改造过程中遗传操作困难。实验室改造过程中菌株生长缓慢,且复杂有机酸较多,增加了理性改造获得高转化率菌株的难度。谷氨酸棒杆菌生产异亮氨酸的策略主要有过表达生物合成途径的关键基因、胞内外转运、解除异亮氨酸对苏氨酸脱水酶和乙酰羟基酸合酶的反馈抑制。研究表明,全局调控基因对异亮氨酸合成途径关键基因具有统筹作用,因此全局调控因子的表达调控对异亮氨酸的生产具有重要作用。
相比于大肠杆菌等工业微生物,能够应用于谷氨酸棒杆菌的遗传操作工具,尤其是在基因组水平编辑的工具较少[13]。因此,从基因组水平上进行单核苷酸突变、基因大片段敲除和插入以及进行基因表达水平调控和外源基因的整合是构建遗传稳定的基础。而外源基因稳定表达工业菌的策略是目前解决这些问题的理想方法,因此许多研究人员致力于开发更高效的基因整合技术[14]。本文综述了广泛应用于谷氨酸棒杆菌基因组整合使用的遗传操作手段,包括同源重组以及较成熟应用于谷氨酸棒杆菌依赖于重组酶的定点整合系统,以期为工业水平稳定生产L-异亮氨酸高产菌株的基因整合策略提供参考依据。
1 L-异亮氨酸的生物合成途径与代谢调控
1.1 L-异亮氨酸的生物合成途径
L-异亮氨酸在谷氨酸棒杆菌的生物合成途径较长,且与其他氨基酸代谢合成途径有重合部分,如苏氨酸、赖氨酸等[15]。谷氨酸棒杆菌以葡萄糖为底物,经过糖酵解途径(embden-meyerhof-parnas pathway,EMP)、戊糖磷酸途径(pentose phosphate pathway,PPP)以及三羧酸循环(tricarboxylic acid cycle,TCA)等中心代谢途径一系列酶催化后生成草酰乙酸,随后在转氨酶作用下生成L-天冬氨酸,经过包括5个限速酶在内的10步酶催化反应,具体生物合成途径如图1所示,最后通过brnFE编码的支链氨基酸胞外转运蛋白将合成的L-异亮氨酸运出胞外。
1.2 L-异亮氨酸的代谢调控
葡萄糖进入胞内,经过L-异亮氨酸生物合成途径合成L-天冬氨酸。L-天冬氨酸首先经过天冬氨酸激酶(aspartate kinase,AK)、天冬氨酸半醛脱氢酶(aspartyl semialdehyde dehydrogenase,ASD)、高丝氨酸脱氢酶(homoserine dehydrogenase,HD)、高丝氨酸激酶(homoserine kinase,HK)和苏氨酸合酶(threonine synthase,TS)催化,并分别由lysc、asd、hom、thrB和thrC基因编码生成苏氨酸;然后经过由苏氨酸脱水酶(threonine dehydrogenase,TD)、乙酰羟基酸合酶(acetohydroxy acid synthase,AHAS)、乙酰羧基酸合成酶(ketol-acid reductoisomerase,AHAIR)、二羟酸还原异构酶(dihydroxy-acid dehydratase,DHAD)和支链氨基酸转氨酶(branched-chain-amino-acid aminotransferase,TA)催化合成L-异亮氨酸,并分别由ilvA、ilvBN、ilvC、ilvD和ilvE编码。L-异亮氨酸的代谢途径有5个关键节点,分别由天冬氨酸激酶、高丝氨酸脱氢酶、高丝氨酸激酶、苏氨酸脱水酶和乙酰羟基酸合成酶5 个关键酶催化。其中天冬氨酸激酶、高丝氨酸脱氢酶和高丝氨酸激酶受到苏氨酸反馈抑制;苏氨酸脱水酶和乙酰羟基酸合成酶受到异亮氨酸的反馈抑制。但在L-苏氨酸到L-异亮氨酸的代谢途径中,酶同样催化L-亮氨酸和L-缬氨酸的生物合成,且乙酰羟基酸合成酶也受到L-亮氨酸和L-缬氨酸的反馈抑制,表明L-异亮氨酸合成过程中代谢调控具有复杂性,具体代谢调控机制如图1所示。

图1 谷氨酸棒杆菌中L-异亮氨酸的生物合成途径及代谢调控机制Fig.1 Biosynthetic pathway and metabolic regulation mechanism of L-isoleucine in Corynebacterium glutamicum
草酰乙酸转氨形成L-天冬氨酸后,从L-天冬氨酸合成L-苏氨酸这部分代谢途径中受到3 个关键酶的催化,同时也是这部分代谢途径的限速步骤,分别是天冬氨酸激酶、高丝氨酸脱氢酶和高丝氨酸激酶。其中天冬氨酸激酶是L-天冬氨酸到L-异亮氨酸生物合成途径的第一个关键节点,催化天冬氨酸到天冬氨酸半醛,催化过程需要消耗ATP,同时该酶也受到L-赖氨酸的反馈抑制[16];高丝氨酸脱氢酶以NADPH 为还原力将天冬氨酸-β-半醛脱羧催化为L-高丝氨酸,在对苏氨酸的反馈抑制中以变构非竞争性方式被抑制;高丝氨酸激酶与高丝氨酸脱氢酶不同,其通过竞争性抑制机制被抑制[17],将L-高丝氨酸催化成为高丝氨酸磷酸。
L-苏氨酸到L-异亮氨酸合成代谢途径中受到2 个关键酶的催化,分别是苏氨酸脱水酶和乙酰羟基酸合成酶,这2 个酶催化的反应不但受到L-异亮氨酸的反馈抑制[18],进而抑制TD 和AHAS的活性,且TD 催化苏氨酸脱水形成2-酮丁酸,AHAS对2-酮丁酸的亲和力大于丙酮酸,2-酮丁酸存在时倾向于合成L-异亮氨酸[19],因此苏氨酸脱水酶催化形成的2-酮丁酸是下一步乙酰羟基酸合成酶催化形成L-异亮氨酸的重要前体;同时AHAS 不但催化1 分子丙酮酸和1 分子酮酸形成异亮氨酸的前体乙酰羟基丁酸[20],而且催化2 分子丙酮酸脱羧形成L-缬氨酸和L-亮氨酸的前体2-乙酰乳酸。因此苏氨酸脱水酶和乙酰羟基酸合成酶在L-异亮氨酸合成过程中不但是限速步骤,而且决定了最终节点处的代谢流向。
2 L-异亮氨酸的生产策略
如图2 所示,根据L-异亮氨酸的生物合成途径和代谢调控,生产L-异亮氨酸的策略主要为:①解除反馈抑制;②过表达L-异亮氨酸合成途径关键基因;③修饰转运系统;④辅助因子平衡。
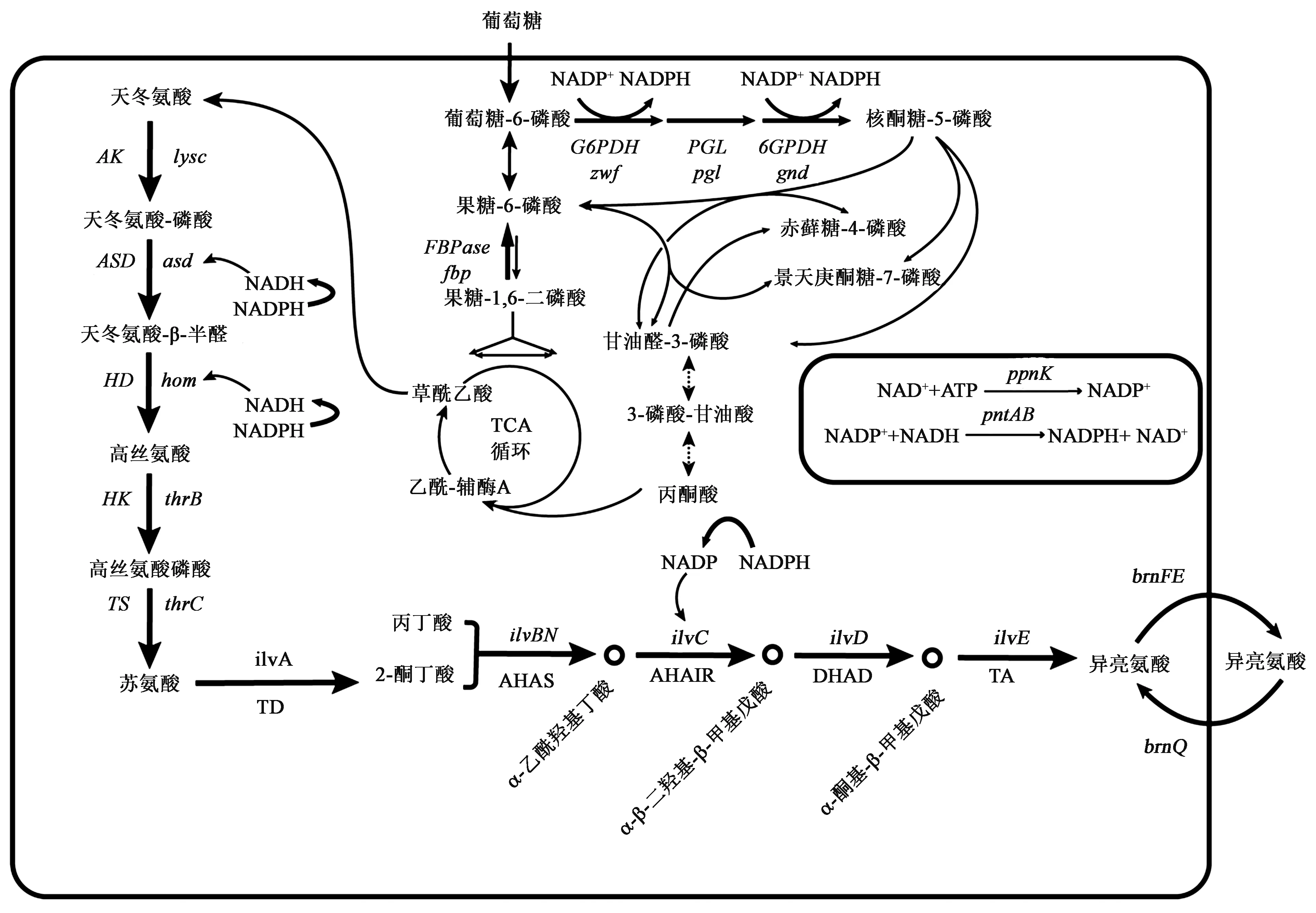
图2 谷氨酸棒杆菌中L-异亮氨酸的改造策略Fig.2 Transformation strategy of L-isoleucine in Corynebacterium glutamicum
2.1 解除反馈抑制
L-异亮氨酸合成过程中由于受到自身的反馈抑制,不但削弱了L-异亮氨酸代谢途径前体的积累,而且导致仅通过过表达基因等增强L-异亮氨酸代谢流强度这一常规代谢工程手段不适用于L-异亮氨酸这种合成途径较长,同时受到多级调控和反馈阻遏的产物。因此通过随机诱变筛选或者定向诱变得到的解除反馈抑制的酶催化作用下能显著提高L-异亮氨酸的产量,对于随机诱变的生产菌株,在进行理性代谢工程改造时可以通过引入解除反馈抑制的突变位点,进而解除L-异亮氨酸对关键节点酶的反馈抑制,最终显著提高了氨基酸产量。
2.1.1 解除L-苏氨酸反馈抑制 L-异亮氨酸生物合成途径不仅包括L-苏氨酸合成途径,也包括L-苏氨酸对天冬氨酸激酶、高丝氨酸脱氢酶和高丝氨酸激酶的反馈抑制,通过构建这3 个关键酶的突变体或者通过对随机诱变得到的高产菌株中编码酶的突变基因进行挖掘,能够得到解除L-苏氨酸反馈抑制的酶突变体,增强L-异亮氨酸合成通路的代谢流。
L-苏氨酸、L-赖氨酸和L-异亮氨酸是天冬氨酸的下游产物,天冬氨酸激酶是天冬氨酸合成L-异亮氨酸的第一个关键酶,L-赖氨酸是天冬氨酸-β-半醛的下游氨基酸,对天冬氨酸激酶有反馈抑制。通过构建天冬氨酸激酶突变体,不仅能不同程度地分别解除L-苏氨酸的反馈抑制,如lysCN374T、lysCE278V和lysCD274A等[21-22],如lysCS381F能够解除L-赖氨酸的反馈抑制[23],有些突变体如lysCT31I和lysCA279T能够解除L-苏氨酸和L-赖氨酸的协同反馈抑制[24-25];高丝氨酸脱氢酶中第378 位突变是解除反馈抑制的重要位点,homG378E和homG378S突变体不同程度解除了L-苏氨酸反馈抑制[25-26];高丝氨酸激酶突变体thrBA20G不但保留了野生酶活性,同时显著降低了L-苏氨酸对该酶的反馈抑制作用[27](表1)。

表1 L-异亮氨酸生物合成途径解除反馈抑制位点Table 1 Feedback inhibition sites of L-isoleucine biosynthesis pathway
2.1.2 解除L-异亮氨酸反馈抑制 从L-苏氨酸到L-异亮氨酸生物合成过程中,苏氨酸脱水酶和乙酰羟基酸合成酶不但是L-异亮氨酸合成的限速步骤,而且对L-异亮氨酸前体合成以及代谢流向具有决定作用,通过引入解除L-异亮氨酸反馈抑制的关键酶,能使代谢流更多流向L-异亮氨酸合成。
苏氨酸脱水酶是L-苏氨酸到L-异亮氨酸代谢途径的第一个关键酶,催化L-苏氨酸脱水形成2-酮丁酸,同时乙酰羟基酸合成酶对2-酮丁酸的亲和力大于丙酮酸,2-酮丁酸存在时倾向于合成L-异亮氨酸[19],因此苏氨酸脱水酶和乙酰羟基酸合成酶共同决定了生成的支链氨基酸种类。解除L-异亮氨酸对苏氨酸脱水酶和支链氨基酸对乙酰羟基酸合成酶的反馈抑制对于L-异亮氨酸的积累具有重要意义,因此通过引入解除反馈抑制的突变位点可提高L-异亮氨酸的产量。苏氨酸脱水酶突变体ilvAV323A和ilvAH278R-L351S解除了L-异亮氨酸对苏氨酸脱水酶的反馈抑制[28];ilvAV140M相比于野生型,苏氨酸脱水酶活性提高了1.5倍,ilvAF383A完全解除了反馈抑制,双突变体ilvAV140M-F383V不但酶活提高了1.5倍,并且完全解除了L-异亮氨酸的反馈抑制[18]。乙酰羟基酸合成酶调节亚基上G20D-I21D-I22F 突变解除了L-缬氨酸、L-异亮氨酸和L-亮氨酸的反馈抑制[29];Pro176Ser、Asp426Glu和Leu575Trp突变后不再受L-异亮氨酸的反馈抑制,L-异亮氨酸的产量有所提高。有研究通过将苏氨酸脱水酶突变体ilvAF383V和乙酰羟基酸合成酶大亚基突变体ilvBNP176S-D426E-L575W共表达后摇瓶发酵,72 h后L-异亮氨酸的产量为30.7 g·L-1,产量提高了131.7%[30](表1)。
2.2 过表达代谢途径关键基因
通过组合过表达L-异亮氨酸合成途径关键基因,主要包括编码5 个重要节点酶、NADPH 的供给相关酶和L-异亮氨酸胞外转运蛋白的基因,其中过表达5 个重要节点酶最主要的是解除L-苏氨酸和L-异亮氨酸反馈抑制的突变体基因,从而加强L-异亮氨酸主代谢流,提高L-异亮氨酸产量。
有研究将ilvBN、ppnK、lrp和brnFE进行不同组合过表达,其中WM005/pYCW-1-ilvBN2-ppnK1在72 h 分批补料发酵后生产32.1 g·L-1的L-异亮氨酸,较对照菌株提高了34.3%[31];fusA编码的核糖体延伸因子G 促进细菌蛋白质合成的易位步骤,frr编码的核糖体循环因子与延伸因子G 共同在翻译终止后将核糖体从信使RNA 中解离,fusA和frr的过表达能够提高谷氨酸棒杆菌IWJ001 中L-异亮氨酸产量,usA和frr以及ilvA、ilvB、ilvN和ppnk基因共同过表达后,L-异亮氨酸产量增加76.5%,并在72 h 补料分批发酵中产生了28.5 g·L-1L-异亮氨酸[32]。
研究表明,除了对谷氨酸棒杆菌胞内L-异亮氨酸合成途径关键酶的过表达,在谷氨酸棒杆菌中过表达大肠杆菌thrABC基因(编码双功能的天冬氨酸激酶Ⅰ-高丝氨酸脱氢酶Ⅰ、高丝氨酸激酶和苏氨酸合成酶),同时敲除alaT(编码丙氨酸氨基转移酶),经过组合这两种改造策略的菌株YILWΔalaT比出发菌株L-异亮氨酸产量提高了17.6%,同时减少了其他副产物氨基酸的积累量,使碳代谢流向L-异亮氨酸生物合成[33]。
2.3 修饰转运系统
由brnF和brnE双组分编码的支链氨基酸胞外转运蛋白能够非特异性向胞外转运支链氨基酸(L-亮氨酸、L-异亮氨酸和L-缬氨酸)和蛋氨酸,并且受到全局调控因子lrp的调节[34]。brnFE编码的胞外转运能够减轻胞内L-异亮氨酸的浓度,减轻合成途径中关键节点的反馈抑制,从而增加L-异亮氨酸的生物合成[35]。brnQ编码支链氨基酸向胞内转运的蛋白。支链氨基酸的胞内转运是由Na+介导[36-37]、质子驱动的[38]。brnFE的过表达提高了L-异亮氨酸的胞外转运效率,brnQ的敲除降低了谷氨酸棒杆菌对胞外L-异亮氨酸的摄取,L-异亮氨酸的净排放量是L-异亮氨酸高产量的重要因素[39]。
谷氨酸棒杆菌中编码支链氨基酸胞外转运蛋白基因brnFE的上调提高了包括L-异亮氨酸在内的多种氨基酸的高效生物合成[40-41]。当胞内支链氨基酸和蛋氨酸积累时,lrp能够结合到lrp与brnF之间的基因区域,保证LRP 在其底物氨基酸积累时才合成[9],进而激活brnFE的表达[42-43]。研究表明lrp、brnFE和lrp-brnFE分别在谷氨酸棒杆菌L-异亮氨酸生产菌中过表达,过表达lrp比过表达brnFE能产生更多的L-异亮氨酸,共同过表达时产量与对照菌相比显著增加,JHI3-156/pDXW-8-lrp-brnFE在摇瓶发酵和分批补料培养条件下L-异亮氨酸产量分别提高了63%和72%[34]。
敲除brnQ基因能够阻断谷氨酸棒杆菌对胞外L-异亮氨酸的摄取,提高L-异亮氨酸的产量[31]。有研究通过降低谷氨酸棒杆菌L-异亮氨酸向胞内摄取和提高L-异亮氨酸向胞外转运,并构建了YILWΔbrnQ、YILWpXMJ19brnFE和YILWΔbrnQ/pXMJ19brnFE,结果表明,与出发菌株相比,L-异亮氨酸外排率均提高,L-异亮氨酸产量分别提高了10.4%、28.2%和43.6%[39]。以上研究表明将降低摄取提高转运这一策略应用于理性改造工业水平的发酵菌株,可提高产量和经济效益。
2.4 辅助因子平衡
NADPH/NADP+影响了细胞内碳流分配和酶催化活性,因此,NADPH 的再生能够维持细胞内依赖NADPH 的代谢活动与酶催化过程。同时代谢工程改造氨基酸高产菌株的关键在于改善胞内NADPH 的供应。因此,通过选择合适的NADPH再生策略能够使碳代谢流最大化流向目标氨基酸。此外,工业菌株可能在生长、生产等发酵阶段需要不同的辅因子水平。如将来自糖酵解的碳通量引入磷酸戊糖途径以增加NADPH 含量有利于目标代谢产物的生成。来自谷氨酸棒杆菌ATCC 13032 的ASD 是一种依赖于NADPH 的酶,需要大量NADPH 才能进行高水平赖氨酸生物合成,NADPH/NADP+的不平衡导致菌株生长状况差,目标产物水平降低[44-45]。
2.4.1 提高NADPH/NADP+的比率 提高NADPH/NADP+的比率主要通过两种策略实现,一种是通过戊糖磷酸途径中基因编码的酶催化作用下将NADP+还原为NADPH;另一种是通过表达编码NAD+激酶的ppnK基因(磷酸化NADH 产生NADPH)。谷氨酸棒杆菌中通过过表达与戊糖磷酸途径(PPP 途径)相关的基因,包括gnd、fbp和pgl,经过分别过表达和组合表达这3个基因,相比于对照菌株,胞内NADPH/NADH+比率提高了3~4 倍,并且生物量和转化率均有明显的提高,其中IWJ001/pDXW-8-gnd-fbp和IWJ001/pDXW-8-gndfbp-pgl相比于对照菌株L-异亮氨酸,产量分别提高了38.4%和65.1%。表明将碳通量引向戊糖磷酸途径能够增加NADPH 的供应,提高胞内NADPH/NADH+比率,进而增加L-异亮氨酸的产量[46]。研究发现,增强zwf、gnd、ppnK、pntAB和udhA基因表达均能提高异亮氨酸的浓度,过表达编码葡萄糖-6-磷酸脱氢酶的zwf基因表现最好,NADPH 和NADPH/NADH+比率分别提高了61.24%和90.63%[47]。将谷氨酸棒杆菌编码NAD+激酶的ppnK基因(磷酸化NAD+产生NADP+)、编码葡萄糖-6-磷酸脱氢酶的zwf基因和来自酿酒酵母编码NADH 激酶的Pos5基因分别过表达,以及zwf-ppnK的共同过表达,均增加了胞内NADPH/NADH+比率和最终L-异亮氨酸的产量。其中zwf-ppnK的共同过表达显著提高了NADPH/NADH+比率,并且使L-异亮氨酸的产量提高了85.9%[48]。
2.4.2 NADPH 再生 构建低成本和高效的NADPH 再生系统不但能保持胞内氧化还原平衡,并且可改善依赖NADPH 产物的产量。NAD+可以通过NAD+激酶转换为NADP+[49],膜连接转氢酶(membrane-linked hydrogenase,PntAB)或可溶性转氢酶(soluble transhydrogenase,UdhA)可以催化NADH 向NADPH 的转化[50],增加转氢酶的活性,并将多余的NADH 转化为NADPH。通过将来自大肠杆菌JM109 菌株中编码pntAB基因和谷氨酸棒杆菌编码NAD+激酶的ppnK基因(催化NADP+到NADPH)共同过表达,显著提高了精氨酸的产量;在大肠杆菌生产木糖醇[51]和3-羟丙酸[52]的研究中,PntAB 增加了两种产物的产量,并且L-异亮氨酸的生产需要更多NADPH[53]。同时也有研究表明NAD+激酶的过表达对异丁醇的生产无显著影响,但NAD+激酶和转氢酶PntAB 的组合会导致异丁醇产量提高,进一步表明转氢酶和NAD+激酶对增加NADPH供应有协同效应[54]。
2.4.3 引入外源依赖NADH的酶 天冬氨酸脱氢酶和天冬氨酸-β-半醛脱氢酶是L-天冬氨酸通路中利用NADPH 的酶,通过在谷氨酸棒杆菌中引入外源依赖NADH 的酶,或者通过突变酶结合中心关键残基改变酶的辅因子偏好从而利用NADH,提高了NADPH/NADH比率,缓解了NADPH供应压力。
引入外源依赖NADH的酶可提高谷氨酸棒杆菌L-赖氨酸产量,已知合成1 mol L-赖氨酸需要4 mol 的NADPH,因此,在L-赖氨酸合成途径引入中直接利用NADH 的酶,有利于L-赖氨酸生产过程中辅助因子平衡。利用酶挖掘技术已经鉴定了一系列利用脱氢酶的NADH,分别为来自铜绿假单胞菌的天冬氨酸脱氢酶(Pseudomonas aeruginosaaspartate dehydrogenase,PaASPDH)、来自运动替斯崔纳菌的天冬氨酸-β-半醛脱氢酶(Tistrella mobilisaspartate-β-semialdehyde dehydrogenase,TmASADH)、来自大肠杆菌的二氢二癸酸还原酶(Escherichia colidihydrodipicolinate reductase,EcDHDPR)和来自假热热孢菌的二氨基庚二酸脱氢酶(Pseudothermotoga thermarumdiaminopimelate dehydrogenase,PtDAPDH)。过 表 达PaASPDH、TmASADH、EcDHDPR 和PtDAPDH 使赖氨酸的产生分别增加了30.7%、32.4%、17.4% 和36.8%。PaASPDH、TmASADH 和EcDHDPR 的组合表达菌株中L-赖氨酸产量最高增加30.7%,过表达4 种NADH 利用酶可以提高L-赖氨酸产量,同时不依赖氧化戊糖磷酸途径提供NADPH[55]。
通过对天冬氨酸-β-半醛脱氢酶结合中心关键残基突变,能够增强辅助因子NAD(H)的利用[56]。EcASADH-Q350N 和 EcASADH-Q350N/H171A突变体在NADH存在时可有效合成L-高丝氨酸;相比野生型,EcASADH 在NADH 作为辅助因子的情况下生产的L-高丝氨酸较少。各策略应用于谷氨酸棒杆菌改造生产L-异亮氨酸的发酵产量见表2。
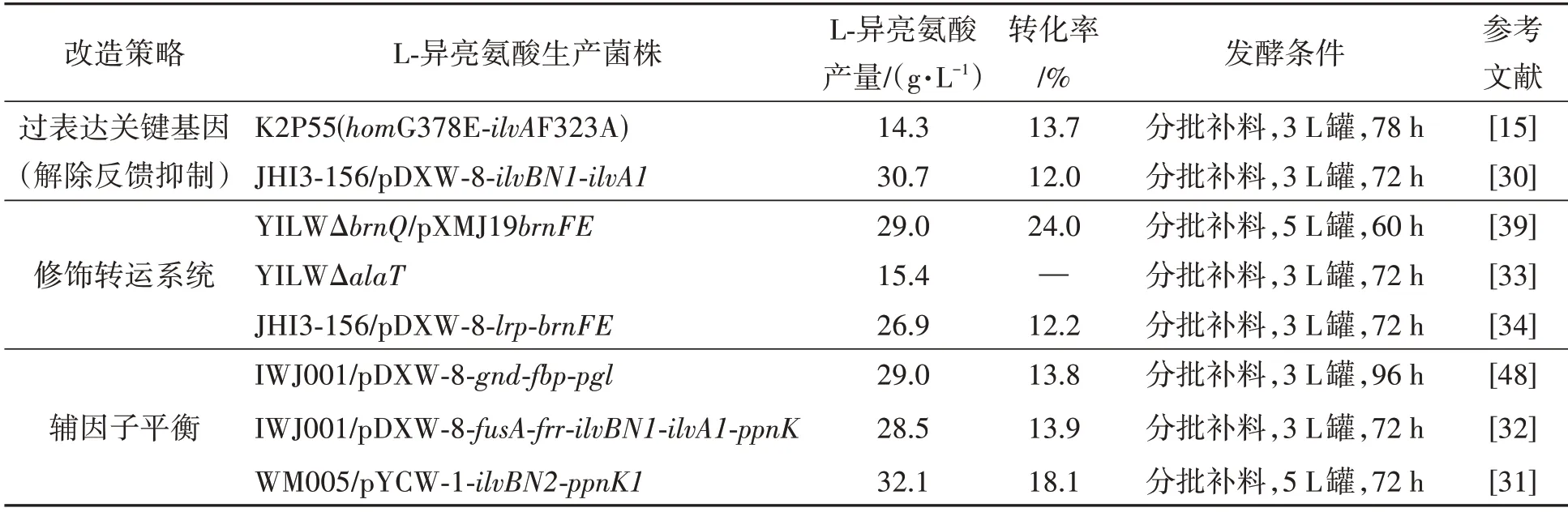
表2 L-异亮氨酸各改造策略发酵产量Table 2 Fermentation yield of L-isoleucine by different transformation strategies
3 基因组整合
一般对于基因表达量的提高和外源基因的表达均是构建于质粒载体上的[57],目前也开发了一系列能应用于谷氨酸棒杆菌的遗传操作工具,如pXMJ19、pECXK99E 和pDXW-8[58]等。质粒表达系统对于菌株的实验室改造具有操作成熟、能实现较高表达量等优势,但应用于工业上大规模生产的菌株则表现出显著的缺点。工业化生产过程中,由于质粒的不稳定性,传代过程中可能造成含质粒菌株表达基因拷贝数的波动和质粒丢失等问题,为了维持质粒稳定性和诱导表达需要添加大量抗生素和诱导剂,这不但降低了工业化发酵生产氨基酸等化学产品的经济效益[57],而且抗生素的大规模应用会严重污染环境[59]。
3.1 自杀质粒介导同源重组
传统基因组整合方式主要以同源重组的方式经过2 次同源重组和2 次筛选验证得到整合目的基因菌株,但是由于载体的转化效率低、第一次同源重组效率低、依赖于抗生素标记基因的使用[60]和蔗糖筛选效率低等问题,需开发新的基因组整合工具。
一般谷氨酸棒杆菌的无痕敲除和基因组整合以pK18mobsacB、pK19mobsacB[61]和pK-JL[62]等成熟应用于谷氨酸棒杆菌敲除的自杀质粒介导。pK18mobsacB 和pK19mobsacB 质粒上带有卡那霉素抗性基因kan和编码果聚糖蔗糖酶的sacB基因,蔗糖存在时sacB基因对谷氨酸棒杆菌具有致死作用,因此sacB基因作为反向筛选标记,经过2次同源重组和2 次筛选后得到目的菌株,但主要用于基因的敲除与交换[63]。随后有研究证明pK18mobsacB 能够在谷氨酸棒杆菌基因组水平上实现基因的过表达[57]。pK-JL 质粒是pK18mobsacB 改良后的自杀质粒,通过替换sacB基因前的启动子为强启动子tacM 后得到的[62],因此提高了该质粒应用于基因组敲除和整合的蔗糖筛选效率,但上述载体仍存在转化效率低、整合质粒构建周期长和第一次同源重组效率低等问题。
通过克隆表达载体pDXW-8 上的Ptac-iMCSrrnBT1T2 元件组装到自杀质粒pK18mobsacB 上,构建了pK18-MBPMT[64],该方法避免了表达质粒的构建,不含有遗传标记,可明显缩短构建整合质粒的时间,尤其对于重复整合等基因组水平过表达操作。同时,对谷氨酸棒杆菌基因组进行无标记特异性位点整合和片段缺失后,可提高L-赖氨酸产量。但该整合质粒仍未解决转化效率低及第一次同源重组效率低的问题。
通过进一步改良谷氨酸棒杆菌中自杀质粒介导的基因编辑系统,将谷氨酸棒杆菌基因组上野生型rspL(编码核糖体小亚基S12 蛋白)突变后获得链霉素抗性,用强启动子Ptuf 表达的野生型rpsL替换pK18mobsacB 中的sacB,因此完成第一次同源重组的菌株能够在链霉素抗性平板生长;随后液体培养,完成第二次同源重组的菌株能够在卡那霉素抗性平板生长,相比pK18mobsacB 蔗糖筛选的30%阳性率,pK18mobrpsL 链霉素筛选阳性率提高至90%,并且由于链霉素和卡那霉素筛选效率较高,可以减少一步法实验步骤和筛选周期,实现双交换[13]。
3.2 Cre/loxP系统
Cre 重组酶(cyclization recombination enzyme)是在大肠杆菌噬菌体P1 中发现的,由Cre基因编码,并由343 个氨基酸残基组成的蛋白质,其能够特异性识别loxP(locus of X-overP1)位点[65],loxP位点长34 bp,包括2个13 bp的反向重复序列和1个8 bp 的间隔区域[66]。反向重复序列是Cre 重组酶特异识别位点,间隔区域决定了loxP位点的方向。Cre/loxP 系统[67]同样利用RecA基因介导的同源重组,经过2 次同源重组后将靶基因替换为两端带有特异重组位点loxP 的kanR 片段,随后通过温敏型质粒表达Cre 重组酶识别特异性重组位点loxP进行重组。2005 年,Suzuki 等[68]开发了基于Cre/loxP 系统对基因组更加精准的敲除方法,删除了谷氨酸棒杆菌基因组上的11 个片段,共有250 kb成功敲除,随后该团队又报道了1 种新的基于Cre/突变loxP 基因组整合系统,其能够将异源基因整合到谷氨酸棒杆菌基因组,但是宿主菌必须含有突变基因,而待整合的外源基因必须包含选择标记,如抗性标记等[14]。之后通过将自杀质粒介导的同源重组与Cre/loxP 系统结合用于谷氨酸棒杆菌基因组敲除,提高了敲除筛选效率[69]。Cre/loxP 系统不仅在原核生物应用广泛,在真核生物生物中也表现出稳定的基因组修饰。利用Cre/loxP 系统介导的同源重组技术失活酿酒酵母中编码亚硫酸盐还原酶的基因MET0提高了生产啤酒的抗氧化能力并改善了风味[70];对耶氏酵母(Yarrowia lipolytica)中编码与γ-癸内酯合成相关的乙酰辅酶A 氧化酶POX5基因用Cre/loxP 同源重组系统进行敲除,成功获得了pox5缺陷菌株。此外,Cre/loxP 系统对哺乳动物细胞,如小鼠[71],由于其具有组织特异性、位点特异性和高效重组等优势,在转基因动物构建中得到了有效利用,同时也是体内外DNA 重组的优势基因操作手段[72]。但经过传统诱变得到的谷氨酸棒杆菌生产菌株时,RecA基因可能会引入非同义突变,甚至可能造成RecA基因表达蛋白的失活,不能介导同源重组。因此开发不依赖于RecA基因介导的重组系统具有重要意义,随着RecT介导的单链重组和人工核酸酶介导的CRISPR的发展,Cre/loxP系统有望更快速、简便、高效地应用于谷氨酸棒杆菌基因组编辑中。
3.3 RecET重组系统
原核生物同源重组大多依赖于内源性的RecA 蛋白,自杀质粒介导的同源重组一般是基于这种构建环状质粒打耙载体实现基因无痕敲除、替换、插入与整合,但构建所需同源臂较长,且筛选过程繁琐。近年来,通过激活原噬菌体Rac 建立的RecET重组系统,成功应用于基因组编辑中,并且不受限制性内切酶切割位点和靶DNA 大小的限制,因此建立了依赖于RecET 介导的基因编辑系统[73]。RecET 系统不仅可以介导双链DNA 重组,其中RecT 也可以介导单链DNA 重组,基于RecT/β 蛋白介导单链重组被用来大规模进化细胞[74],实现了多样自动化的基因组改造技术。
RecET 系统首先在大肠杆菌中应用,通过PCR 扩增得到的DNA 同源片段替换大肠杆菌中的靶基因实现了目的基因的敲除[75]。RecET 通常被构建于质粒上,由于大肠杆菌内源存在recBCD,能够降解外源线性双链DNA,因此,RecET 系统的优化主要集中于抑制recBCD基因功能,保证线性DNA 分子的完整性,从而顺利利用RecET 系统介导双链DNA 重组[76]。随着RecET系统重组技术的发展,研究发现70 bp 长的双链DNA 和单链DNA 均可以作为打靶分子对基因组进行点突变和插入修饰[77]。单链重组的原理是将单链DNA 转入细胞后,在基因组复制过程中,由于基因的半不连续复制,设计的单链DNA 以冈崎片段的形式结合于后随链后在基因组稳定遗传。随着单链重组技术在谷氨酸棒杆菌中的应用,对多种RecT 同源蛋白进行比较优化,发现RecT 介导单链DNA 进行单链重组效果最好[78]。RecT 介导的单链重组也被用以构建基因组多样性文库,并通过构建单链DNA 文库实现基因组多样性[74],但得到的突变体也需要与高通量的筛选技术相结合。
随着基因组编辑技术的迅猛发展,各种基因编辑技术结合实现基因组的高效编辑和高效筛选成为一种基因组改造策略。如将RecET 技术和Cre/loxP 系统结合,实现了对谷氨酸棒杆菌ATCC 14067无标记缺失[79],这也为谷氨酸棒杆菌基因编辑提供了一种无标记操作策略。通过优化CRISPR/Cpf1和RecET系统,实现了对谷氨酸棒杆菌基因组大片段多基因的编辑[80],利用crRNA、CRISPR/Cpf1-RecET 一步可以编辑2~3 个基因,效率分别为91.6%和10%,1、5、20 kb 的编辑效率分别为79.6%、91.3%、36.4%。
3.4 CRISPR/Cas9
规律间隔成簇短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)是指含有较多短的不连续的重复序列(24~37 nt),Cas 是指与CRISPR 相关的蛋白,二者共同作用组成CRISPR-Cas 系统,用以抵御外来因子RNA 介导的获得性免疫系统。CRISPR 前导序列(leader sequence)类似于启动子,富含A/T 低复杂性的非编码区,起始转录重复间隔序列并生成crRNA(CRISPR RNAs)前体,CRISPR 序列往往邻近cas基因,随后间隔区(spacers)识别外来遗传因子,crRNA 与Cas 蛋白共同参与免疫防御过程。来自化脓球杆菌的Cas效应蛋白SpCas9对谷氨酸棒杆菌存在毒性,通过使用来自链霉菌的SpCas9发现其能够在谷氨酸棒杆菌中得到转化子,同时结合单链重组系统删除了谷氨酸棒杆菌基因组400 bp左右,但更大片段的删除、插入以及点突变等尚未测试。由于谷氨酸棒杆菌在工业化生产氨基酸等发酵产品有良好性能,被越来越多的进行改造并用于发酵生产[81],但目前主要对谷氨酸棒杆菌的基因组改造主要依赖于效率极低的同源双交换[82]。CRISPR/Cas9 具有效率高、价格低廉、操作简单等优势,并广泛应用于哺乳动物、植物和微生物中,谷氨酸棒杆菌也开发了基于CRISPR/Cas9 的基因组编辑系统[83],未来有望应用于工业生产。
同时,有研究指出SpCas9 具有毒性,因此使用来自Francisella novicida(FN)的Cas9 效应蛋白(FNCpf1)与谷氨酸棒杆菌适配,同时结合单链重组系统,能够对基因组进行删除、插入等精细修改,明显缩短了基因组编辑周期[84]。有研究在耻垢分枝杆菌中测试了14种不同Cas9效应蛋白,结果发现3 个Cas9基因不影响耻垢分枝杆菌的生长,而且表现出了更高的切割活性[85]。因此,FNCpf1 相比于SpCas9 更适合用于对谷氨酸棒杆菌基因组编辑,并且有更广泛的适用空间。
CRISPR/Cas9 除了直接用于基因组编辑,目前还作为1 种如RecT 介导的单链重组技术和非同源末端连接(non-homologous end joining,NHEJ)的高效筛选策略。RecT 介导的单链重组技术通过几十个碱基作为单链DNA 即可实现基因组编辑,但重组子阳性率较低。通过在谷氨酸棒杆菌中利用CRISPR-Cas9介导突变后重组子作为辅助筛选策略[86],显著提高了筛选效率,减轻工作周期,使RecT介导的单链重组技术能更高效地应用于基因组编辑。随着基因组编辑技术深入地发展以及在谷氨酸棒杆菌中的广泛应用,同时,各个编辑技术之间的协同辅助策略也能够为谷氨酸棒杆菌中的基因组编辑操作提供更高效的工具。
4 展望
L-异亮氨酸作为人体8 种必需氨基酸之一,在食品、药品和化妆品等领域应用广泛,且需求量逐年上升。但工业发酵L-异亮氨酸大部分通过随机诱变得到,菌株稳定性差,目前主流方法为随机诱变后的菌株再进行理性改造。理性改造策略除了针对L-异亮氨酸生物合成途径关键酶的过表达、转运系统、关键酶的催化活性反馈抑制能力外,代谢工程改造氨基酸高产菌株的关键还在于改善胞内NADPH 的供应。传统代谢工程对于氨基酸代谢途径关键基因操作的分析改造非常重要,而且酶挖掘和辅助因子工程相结合也是提高氨基酸产量的高效方法。由于质粒的稳定性差,开发更高效的基因组遗传操作工具尤为重要,随着基因组操作工具水平的提高,以谷氨酸棒杆菌为底盘细胞的发酵生产水平也必将大幅提高。
随着全基因组测序、转录组学、代谢组学等现代生物学技术的发展,以及生物信息学与计算机建模技术与生命科学多学科融合,未来对谷氨酸棒杆菌代谢的改造需从菌株整体代谢网络出发,通过全局分析以及代谢流重构,从而进一步提高谷氨酸棒杆菌生产L-异亮氨酸的产量。
猜你喜欢
河北省科学院学报(2023年4期)2023-08-29 13:10:04
山东畜牧兽医(2023年8期)2023-08-28 08:05:50
食品与发酵工业(2022年5期)2022-03-30 09:02:04
天津科技大学学报(2020年3期)2020-06-23 08:39:00
食品与发酵工业(2019年12期)2019-07-04 03:09:24
山东化工(2017年22期)2017-12-20 02:43:37
饲料工业(2017年8期)2017-04-05 04:43:34
化工设计通讯(2017年10期)2017-03-02 03:24:02
广东饲料(2016年1期)2016-12-01 03:43:01
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:05
