
HPLC法同时测定续断药材中7个成分的含量
2022-03-29 23:35杨昌贵龚安慧张成刚肖承鸿凡迪周涛
中国药房 2022年6期
杨昌贵 龚安慧 张成刚 肖承鸿 凡迪 周涛

中圖分类号 R284.1 文献标志码 A 文章编号 1001-0408(2022)06-0680-05
DOI 10.6039/j.issn.1001-0408.2022.06.06
摘 要 目的 建立同时测定续断药材中6个环烯醚萜类成分(马钱苷酸、马钱子苷、当药苷、续断苷B、续断苷A、林生续断苷Ⅰ)和1个三萜皂苷类成分(川续断皂苷Ⅵ)含量的方法。方法 采用高效液相色谱(HPLC)法。以Symmetry® C18为色谱柱,以乙腈-0.1%磷酸溶液为流动相进行梯度洗脱,检测波长为212 nm(川续断皂苷Ⅵ)、237 nm(续断苷B、续断苷A、当药苷、马钱苷酸、林生续断苷Ⅰ、马钱子苷),柱温为30 ℃,流速为1.0 mL/min,进样量为20 μL。结果 马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ检测质量浓度的线性范围分别为399.24~931.56、50.30~150.90、48.24~168.84、27.00~70.20、12.93~38.80、40.64~121.92、42.08~147.28 µg/mL(r均大于0.999 0);精密度、重复性、稳定性(24 h)试验的RSD均小于2%;平均加样回收率分别为104.43%(RSD=0.63%,n=6)、101.74%(RSD=1.11%,n=6)、100.76%(RSD=1.06%,n=6)、98.00%(RSD=1.58%,n=6)、99.03%(RSD=2.31%,n=6)、102.93%(RSD=2.26%,n=6)、102.31%(RSD=1.00%,n=6);含量分别为142.5~280.6、5.5~49.0、28.0~112.9、7.2~35.8、4.4~16.9、17.2~79.3、0.8~54.5 mg/g。结论 所建含量测定方法准确、可靠,可用于续断药材中7个成分的含量测定。
关键词 续断;含量测定;高效液相色谱法;三萜皂苷;环烯醚萜
Simultaneous determination of 7 components in Dipsacus asper by HPLC
YANG Changgui1,GONG Anhui2,ZHANG Chenggang1,XIAO Chenghong1,FAN Di3,ZHOU Tao1(1. Institute of Traditional Chinese Medicine and Ethnic Medicine Resources, Guizhou University of Traditional Chinese Medicine, Guiyang 550025, China; 2. Dept. of Pharmacy, Guizhou Health Vocational College, Guizhou Tongren 554300, China; 3. Guizhou Crop Technology Promotion Station, Guiyang 550001, China)
ABSTRACT OBJECTIVE To establish the method for the simultaneous determination of six iridoids (loganic acid, loganin, sweroside, dipsanoside B, dipsanoside A, sylvestroside Ⅰ) and one triterpene saponin (asperosaponin Ⅵ) in Dipsacus asper. METHODS High performance liquid chromatography (HPLC) method was adopted. The determination was performed on Symmetry® C18 column with mobile phase consisted of acetonitrile-0.1% phosphoric acid solution (gradient elution) at the flow rate of 1.0 mL/min. The detection wavelengths were set at 212 nm (asperosaponin Ⅵ) and 237 nm (dipsanoside B, dipsanoside A, sweroside, loganic acid, sylvestroside Ⅰ, loganin). The column temperature was set at 30 ℃, and sample size was 20 μL. RESULTS The linear range of loganic acid, loganin, sweroside, sylvestroside Ⅰ, dipsanoside B, dipsanoside A and asperosaponin Ⅵ were 399.24-931.56, 50.30-150.90, 48.24-168.84, 27.00-70.20, 12.93-38.80, 40.64-121.92, 42.08-147.28 µg/mL(all r>0.999 0). RSDs of precision, reproducibility and stability tests (24 h) were all less than 2%. Average recoveries were 104.43%(RSD=0.63%, n=6), 101.74%(RSD=1.11%,n=6), 100.76%(RSD=1.06%,n=6), 98.00%(RSD=1.58%,n=6), 99.03%(RSD=2.31%, n=6), 102.93%(RSD=2.26%,n=6), 102.31%(RSD=1.00%,n=6), respectively, The contents were 142.5-280.6, 5.5-49.0, 28.0-112.9, 7.2-35.8, 4.4-16.9, 17.2-79.3, 0.8-54.5 mg/g, respectively. CONCLUSIONS Established method is accurate and reliable, and can be used for the content determination of 7 components in D. asper.
KEYWORDS Dipsacus asper; content determination; high performance liquid chromatography; triterpene saponins; iridoids
续断为川续断科植物川续断Dipsacus asper Wall. ex Henry的干燥根,现收载于2020年版《中国药典》(一部),具有补肝肾、强筋骨、续折伤、止崩漏的功效,用于治疗肝肾不足、筋伤骨折、胎漏等症[1]。川续断资源丰富,广泛分布于我国贵州、湖北、湖南、江西、广西、云南、四川和西藏等地[2]。有研究表明,续断主要含有三萜皂苷类、环烯醚萜类、生物碱类、挥发油类等成分[3]。其中,三萜皂苷类和环烯醚萜类成分为该药材的主要药效成分[4]。三萜皂苷类成分主要包括川续断皂苷Ⅵ、川续断皂苷Ⅸ、川续断皂苷Ⅹ、川续断皂苷Ⅻ、灰毡毛忍冬皂苷甲、灰毡毛忍冬皂苷乙等[4-5],具有修复和愈合骨损伤的作用[6-7],尤以川续断皂苷Ⅵ的研究较为深入。现代药理研究表明,川续断皂苷Ⅵ可通过抑制软骨基质蛋白多糖分解来降低关节腔内炎症介质的表达,从而阻止关节软骨形态改变,延缓关节退变[8-10]。环烯醚萜类化合物的主要成分为当药苷、马钱苷酸、马钱子苷、续断苷A、续断苷B、林生续断苷Ⅰ等[4],具有促进骨伤愈合、改善学习记忆、提高脑组织抗氧化的作用[10-12]。
2020年版《中国药典》(一部)规定了续断药材中川续断皂苷Ⅵ的含量不得少于2.0%,同时亦规定了水分、总灰分、酸不溶性灰分分别不得过10.0%、12.0%、3.0%,水溶性浸出物不得少于45.0%[1]。此外,有学者对续断的质量进行了评价:冯汪银等[13]以川续断皂苷Ⅵ为质量评价指标,比较了不同等级续断药材的质量差异;魏庆红等[14]以川续断皂苷Ⅵ和总皂苷含量为评价指标,探讨了不同加工工艺对续断药材质量的影响。但上述研究的评价指标较为单一,不能全面表征药材的整体质量;加之续断药材所含药效成分复杂,除川续断皂苷Ⅵ外,马钱苷酸、马钱子苷、当药苷、续断苷A、续断苷B及林生续断苷Ⅰ这6个环烯醚萜类成分在续断药材中的含量也较高,且目前尚未见基于环烯醚萜类成分的质量报道。基于此,本研究拟采用高效液相色谱(high performance liquid chromatography,HPLC)法建立同时测定续断药材中6个环烯醚萜类成分(马钱苷酸、马钱子苷、当药苷、续断苷B、续断苷A及林生续断苷Ⅰ)和1个三萜皂苷类成分(川续断皂苷Ⅵ)含量的方法,旨在为续断药材的质量控制提供参考。
1 材料
1.1 主要仪器
本研究所用主要仪器有e2695型HPLC仪及配备的四元梯度输液泵、二极管阵列检测器、自动进样器和柱温箱(美国Waters公司),ME204型电子分析天平[梅特勒-托利多仪器(上海)有限公司],101-2AB型超声清洗机(天津市泰斯特仪器有限公司)等。
1.2 主要药品与试剂
马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ对照品均由山东省分析测试中心制备,通过核磁共振氢谱(1H nuclear magnetic resonance,1H-NMR)、核磁共振碳谱(13C nuclear magne- tic resonance,13C-NMR)、质谱(mass spectrometry,MS)法验证结构,并经HPLC法检测纯度均大于98%(面积归一化法);乙腈、甲醇均为色谱纯,乙醇、磷酸均为分析纯,水为重蒸馏水。
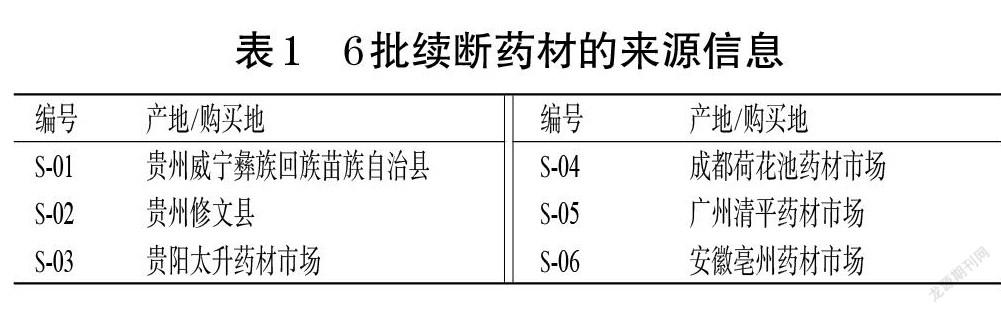
6批续断药材(编号S-01~S-06)经贵州中医药大学药学院江维克教授鉴定为川续断科植物川续断D. asper Wall. ex Henry的干燥根。6批续断药材的来源信息见表1。
2 方法与结果
2.1 色谱条件
以Symmetry® C18(250 mm×4.6 mm,5 μm)为色谱柱,以乙腈(A)-0.1%磷酸溶液(B)为流动相进行梯度洗脱(0~5 min,10%A;5~12 min,10%A→13.3%A;12~23 min,13.3%A;23~45 min,13.3%A→37%A;45~55 min,37%A);檢测波长为212 nm(川续断皂苷Ⅵ)、237 nm(续断苷B、续断苷A、当药苷、马钱苷酸、林生续断苷Ⅰ、马钱子苷);柱温为30 ℃;流速为1.0 mL/min;进样量为20 μL。
2.2 溶液的制备
2.2.1 供试品溶液 取续断药材样品细粉,约0.5 g,精密称定,置于具塞锥形瓶中,精密加入20%乙醇25 mL,密塞,称定质量,浸泡30 min后超声(功率100 W,频率40 kHz)处理30 min,放冷,再次称定质量,用20%乙醇补足减失的质量,摇匀,滤过,精密量取续滤液5 mL,置于25 mL量瓶中,用20%乙醇稀释至刻度,摇匀,即得。
2.2.2 对照品溶液 取各对照品适量,精密称定,用甲醇溶解,制成马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ质量浓度分别为6.654、2.012、2.412、1.080、0.970、2.032、3.156 mg/mL的单一对照品储备液。分别精密吸取上述各单一对照品储备液1.5、0.9、0.8、0.9、0.4、0.8、0.4 mL,置于同一20 mL量瓶中,用甲醇稀释至刻度,摇匀,得马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ质量浓度分别为499.05、90.54、96.48、48.60、19.40、81.28、63.12 µg/mL的混合对照品溶液。
2.2.3 空白溶剂 以20%乙醇为空白溶剂。
2.3 系统适用性试验
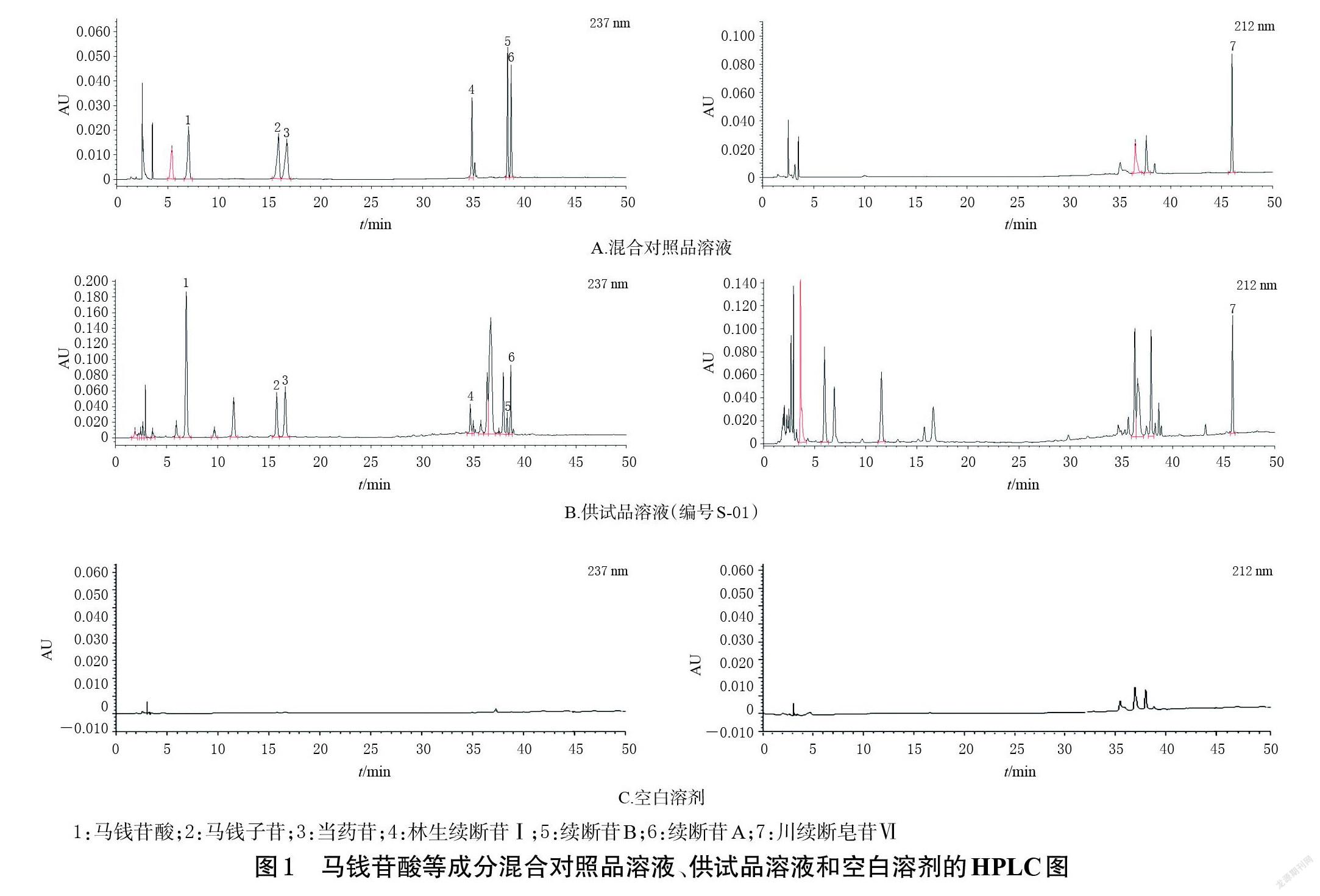
取“2.2”项下混合对照品溶液、供试品溶液、空白溶剂,按“2.1”项下色谱条件进样测定,记录色谱图,详见图1。由图1可知,在该色谱条件下,理论板数按马钱苷酸峰计均不低于5 000,各色谱峰与相邻色谱峰的分离度均大于1.5,各成分测定不受其他组分的干扰,空白溶剂对测定无干扰。
2.4 线性关系考察
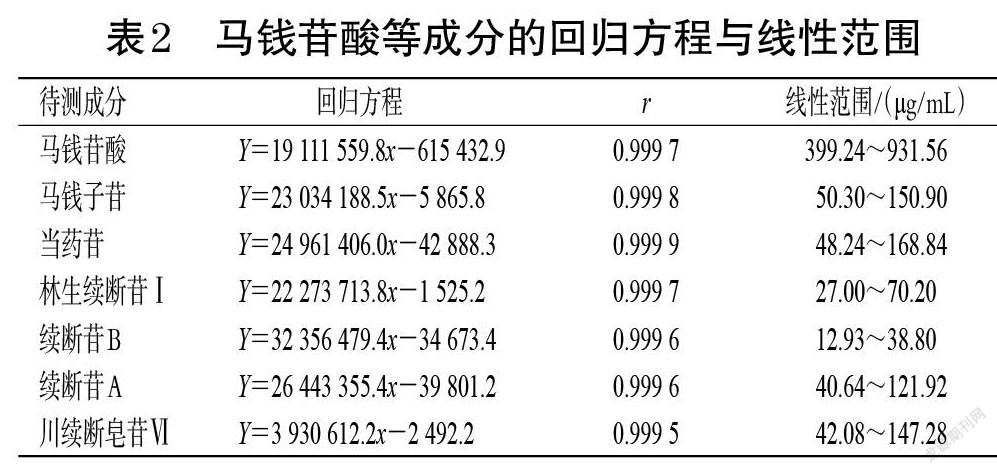
精密吸取“2.2.2”项下马钱苷酸对照品储备液0.9、1.2、1.5、1.8、2.1 mL,置于15 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为399.24、532.32、665.40、798.48、931.56 µg/mL的马钱苷酸系列工作溶液;精密吸取马钱子苷对照品储备液0.5、0.7、0.9、1.1、1.5 mL,置于20 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为50.30、70.42、90.54、110.66、150.90 µg/mL的马钱子苷系列工作溶液;精密吸取当药苷对照品储备液0.4、0.6、0.8、1.0、1.4 mL,置于20 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为48.24、72.36、96.48、120.60、168.84 µg/mL的当药苷系列工作溶液;精密吸取林生续断苷Ⅰ对照品储备液0.5、0.7、0.9、1.1、1.3 mL,置于20 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为27.00、37.80、48.60、59.40、70.20 µg/mL的林生续断苷Ⅰ系列工作溶液;精密吸取续断苷B对照品储备液0.2、0.3、0.4、0.5、0.6 mL,置于15 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为12.93、19.40、25.87、32.33、38.80 µg/mL的续断苷B系列工作溶液;精密吸取续断苷A对照品储备液0.4、0.6、0.8、1.0、1.2 mL,置于20 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为40.64、60.96、81.28、101.60、121.92 µg/mL的续断苷A系列工作溶液;精密吸取川续断皂苷Ⅵ对照品储备液0.2、0.3、0.4、0.5、0.7 mL,置于15 mL量瓶中,用甲醇稀释至刻度,制得质量浓度分别为42.08、63.12、84.16、105.2、147.28 µg/mL的川续断皂苷Ⅵ系列工作溶液。取各工作溶液按“2.1”项下色谱条件进样测定,以各待测成分质量浓度(x)为横坐标、峰面积(Y)为纵坐标进行线性回归,结果见表2。
2.5 精密度试验
取“2.2.2”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果显示,马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ峰面积的RSD分别为0.51%、0.49%、0.44%、0.33%、0.33%、0.47%、0.84%(n=6),表明仪器精密度良好。
2.6 稳定性试验
取“2.2.1”项下供试品溶液(编号S-01),分别于室温下放置0、2、4、6、12、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果显示,马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ峰面积的RSD分别为0.35%、1.56%、1.18%、1.89%、0.72%、1.94%、1.52%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.7 重复性试验
精密称取续断药材(编号S-01)粉末,每份约0.5 g,共6份,按“2.2.1”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品含量。结果显示,马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ含量的RSD分别为1.20%、1.70%、1.78%、1.58%、1.78%、0.44%、1.36%(n=6),表明方法重復性良好。
2.8 加样回收率试验
取已知含量的续断药材(编号S-01)粉末,每份约0.25 g,共6份,精密称定,置于100 mL锥形瓶中,分别精密加入“2.2.2”项下各单一对照品储备液5.5、2.6、2.9、1.6、1.1、2.1、2.6 mL,按“2.2.1”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表3。
2.9 样品含量测定
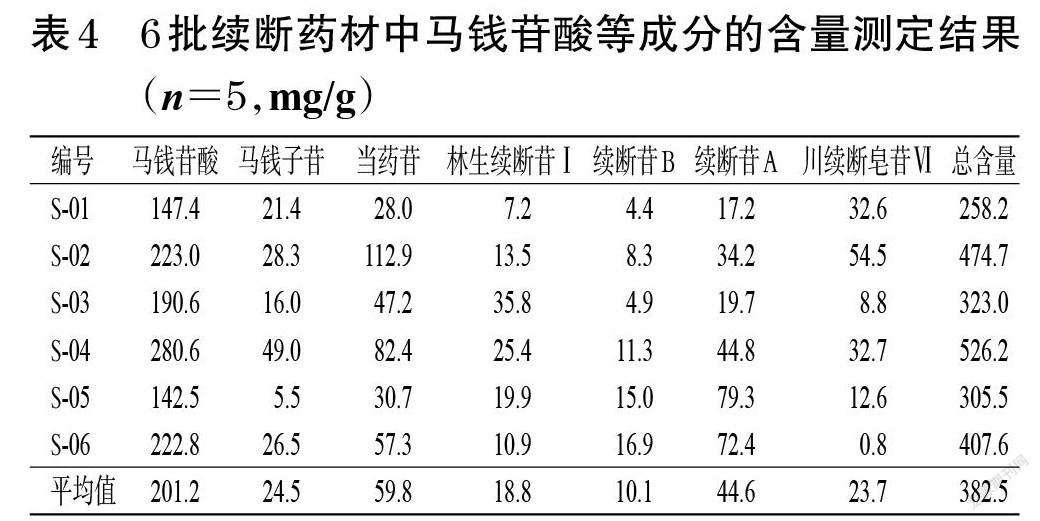
精密称取6批续断药材粉末,各约0.5 g,按“2.2.1”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品含量(质量浓度若超出线性范围,则适当稀释或浓缩)。每样品平行操作5次,取平均值,结果见表4。
3 讨论
本课题组采用二极管阵列检测器在190~800 nm波长范围内对7个待测成分进行扫描后发现,马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ的最大吸收波长分别为236、238、246、237、236、237、212 nm;为了检测方便并能有效反映各成分含量,暂定川续断皂苷Ⅵ的检测波长为212 nm;马钱苷酸等其余6个成分的检测波长为237 nm。预实验结果显示,与在相应最大吸收波长条件下所得结果比较,在上述波长条件下所得各成分含量均无显著差异,故选择川续断皂苷Ⅵ的检测波长为212 nm,马钱苷酸等其余6个成分的检测波长为237 nm。系统适用性试验结果显示,在该条件下,各色谱峰分离度良好、基线平稳。同时,本课题组考察了乙腈-水、乙腈-0.1%磷酸溶液2种流动相体系的分离效果。结果显示,以乙腈-水为流动相时,续断苷B、续断苷A的色谱峰峰形欠佳且分离度较差;以乙腈-0.1%磷酸溶液为流动相时,马钱苷酸等7个成分均能较好地分离,且出峰时间稳定,故选择乙腈-0.1%磷酸溶液为流动相。此外,本课题组前期通过单因素实验比较了不同体积分数(10%、20%、30%、40%、50%、75%、95%)乙醇超声和回流的提取效果及对方法学考察的影响。结果显示,当以10%乙醇为溶剂时,方法的重复性较差;当以20%乙醇为溶剂时,马钱苷酸等7个成分含量和方法稳定性均较优。同时,笔者发现,超声提取和回流提取的效果相当,从操作的简便性和方法的重复性考虑,本研究选择超声提取。在此基础上,本课题组还考察了不同超声处理方式(浸泡30 min后超声处理、直接超声处理)、提取时间(50、40、30、20 min)对提取效果的影响。结果发现,将药材浸泡30 min后再超声处理的提取率和方法的重复性均优于直接超声处理;提取20~50 min时,马钱苷酸等7个成分的含量无显著差异。综合考虑,本研究选择20%乙醇为溶剂,浸泡30 min后超声提取30 min。
本研究结果显示,马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A、川续断皂苷Ⅵ的含量分别为142.5~280.6、5.5~49.0、28.0~112.9、7.2~35.8、4.4~16.9、17.2~79.3、0.8~54.5 mg/g,提示7个成分含量存在较大差异,平均含量最高的为马钱苷酸(201.2 mg/g),最低的为续断苷B(10.1 mg/g),二者平均含量相差近20倍。从7个成分的总量来看,总含量最高的为成都荷花池药材市场的样品(S-04,526.2 mg/g),最低的为贵州威宁彝族回族苗族自治县的样品(S-01,258.2 mg/g),含量相差近2倍。不同批次药材间7个成分的含量亦存在显著差异,如从马钱苷酸含量来看,成都荷花池药材市场样品的含量最高(S-04,280.6 mg/g),广州清平药材市场样品的含量最低(S-05,142.5 mg/g),二者含量相差近2倍,其原因可能与样品的产地环境和加工炮制方式有关。有研究表明,不同产地续断药材中川续断皂苷Ⅵ含量差异较大,可达10倍以上[15],其含量与产地海拔呈正相关[16];此外,不同炮制方法会影响续断中马钱苷酸、川续断皂苷乙、川续断皂苷Ⅵ、绿原酸等成分的含量,加工方式的不同可造成川续断皂苷乙、绿原酸成分含量相差近20倍[17-18]。
中药成分复杂,其药效的发挥往往是多个成分协同作用的结果,若仅以一、两个成分作为质量评价指标难以反映药材的真实质量,而采用多指标评价的方法可更全面地表征药材质量,故多成分的含量测定已成为中药材质量评价的发展趋势[19]。本研究所建续断药材中马钱苷酸等7个成分含量的检测方法,不仅包括了2020年版《中国药典》(一部)规定的指标成分川续断皂苷Ⅵ的测定,同时还对具有药理活性的6个环烯醚萜类成分(马钱苷酸、马钱子苷、当药苷、林生续断苷Ⅰ、续断苷B、续断苷A)进行了同时检测,可以更加全面地反映续断药材的内在质量。
综上所述,本研究所建含量测定方法准确、可靠,可用于续断药材中7个成分的含量测定。
参考文献
[ 1 ] 国家药典委员会.中华人民共和国药典:一部[S]. 2020年版.北京:中国医药科技出版社,2020:343-344.
[ 2 ] 中国科学院中国植物志编辑委员会.中国植物志:第七十三卷:第一分册[M].北京:科学出版社,1986:63.
[ 3 ] 刘二伟,吴帅,樊官伟.川续断化学成分及药理作用研究进展[J].中华中医药学刊,2010,28(7):1421-1423.
[ 4 ] 刘京晶.续断生药学研究[D].北京:北京协和医学院,2011.
[ 5 ] 张永文,薛智.川续断中皂甙(苷)Ⅺ,Ⅻ和ⅩⅢ的结构研究[J].药学学报,1993,28(5):358-363.
[ 6 ] 李贤让.续断总皂苷治疗骨关节炎及其机制的实验研究[D].济南:山东大学,2017.
[ 7 ] LIU K F,LIU Y,XU Y T,et al. Asperosaponin Ⅵ protects against bone destructions in collagen induced arthritis by inhibiting osteoclastogenesis[J]. Phytomedicine,2019,63:153006.
[ 8 ] GU M B,JIN J,REN C H,et al. Akebia saponin D suppresses inflammation in chondrocytes via the NRF2/HO-1/NF-κB axis and ameliorates osteoarthritis in mice[J]. Food Funct,2020,11(12):10852-10863.
[ 9 ] GONG L L,YANG S,LIU H,et al. Anti-nociceptive and anti-inflammatory potentials of akebia saponin D[J]. Eur J Pharmacol,2019,845:85-90.
[10] 杨延平,杨勇.续断抗骨质疏松活性部位的筛选[J].今日药学,2012,22(6):342-344.
[11] 匡海学.中药化学[M].北京:中国中医药出版社,2003:189.
[12] 何雪心,裘军.续断提取物对小鼠D-半乳糖拟痴呆模型的抗氧化作用[J].中国药师,2005,8(3):185-187.
[13] 冯汪银,周涛,肖承鸿,等.续断商品规格等级标准及质量评价研究[J].中国中药杂志,2019,44(14):2996-3001.
[14] 魏庆红,胡雨,金传山,等.不同加工工艺对续断质量的影响[J].中国药业,2015,24(18):14-16.
[15] 冯良.不同产地续断中4种皂苷类成分的HPLC含量测定[J].社区医学杂志,2017,15(13):50-52.
[16] 江维克,艾强,周涛,等.贵州续断药材川续断皂苷Ⅵ含量的地理分布趋势分析[J].贵州农业科学,2013,41(8):19-22.
[17] 张祺嘉钰,孙毅,赵重博,等.不同炮制方法对不同产地续断中有效成分含量及抗氧化活性的影响[J].西北药学杂志,2020,35(2):169-172.
[18] 严福林,魏升华,王志威,等. HPLC法同時测定不同加工续断药材中2种成分[J].贵州中医药大学学报,2021,43(3):34-39.
[19] 姚静,孙欣光,董蓉,等. HPLC-CAD一测多评法同时测定黄芪中6种成分含量[J].药学学报,2021,56(2):557- 564.
(收稿日期:2021-11-08 修回日期:2022-02-05)
(编辑:陈 宏)
猜你喜欢
中国当代医药(2016年30期)2017-01-07
中国当代医药(2016年29期)2017-01-03
中国医药导报(2016年30期)2016-12-28
云南中医中药杂志(2016年11期)2016-12-26
中国民族民间医药·上半月(2016年11期)2016-12-26
中国民族民间医药·上半月(2016年11期)2016-12-26
考试周刊(2016年95期)2016-12-21
