
VOCs燃烧催化剂耐硫性新进展
2022-03-25 03:25吴冬霞胡江亮侯建材常丽萍王建成鲍卫仁
洁净煤技术 2022年2期
吴冬霞,程 行,胡江亮,侯建材,常丽萍,王建成,鲍卫仁
(1.太原理工大学 煤科学与技术教育部重点实验室,山西 太原 030024;2.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
0 引 言
挥发性有机化合物(VOCs)是在常温常压下沸点≤250 ℃的有机化合物,主要包括烷烃类、芳香烃类、烯烃类、卤烃类、酯类、醛类、酮类等[1]。VOCs不仅对人体有明显的毒性效应,还对环境有多重危害[2-3]。山西省是我国最重要的焦炭生产基地,2019年焦炭产量9 699万t,占全国总产量的20.6%,居全国首位。独特的产业特点使焦化行业成为重要的VOCs排放源。随着国家“十四五”PM2.5和O3协同控制规划与山西省焦化行业超低排放改造方案的实施,PM2.5和O3等区域大气复合污染物防控成为目前该区域大气污染防治的核心工作。VOCs作为PM2.5和O3共同的关键前体物,对2者协同控制具有重要意义,加强VOCs的综合治理迫在眉睫。
目前,山西大多数焦化厂,除焦炉生产过程外,已基本实现无组织排放VOCs废气的有效收集,将无组织逸散变为有组织排放,因此如何安全有效处理这些有组织废气成为当务之急。国内针对其治理已有一定的研发基础,形成了以末端治理为核心的多种焦化VOCs治理手段。主流应用技术包括吸收、吸附、光催化降解、生物降解、返燃烧室直接燃烧、催化燃烧等[4],综合目前应用情况分析,除污水单元外,其他焦化工段建立以燃烧为核心的治理技术将成为未来趋势。其中催化燃烧适合处理大风量、低浓度VOCs废气,且能耗低、不副产污染物,具有巨大的应用潜力。但在实际工业应用中,由于煤中硫原子的迁移,焦化VOCs废气多含有硫组分,SO2和H2S浓度最高。这些硫化物会对焦化VOCs燃烧催化剂活性产生极大影响。通常,硫化物体积分数超过5×10-6时,其催化活性受到抑制[5-6]。硫化物不仅会与VOCs分子竞争吸附活性位点,还会与催化剂组分作用生成硫酸盐覆盖在催化剂表面,破坏催化剂结构,减少活性位点,致使催化剂中毒,抑制催化活性。因此,催化燃烧技术面临着设计开发耐硫催化剂的应用难题,硫化物对催化剂的影响已成为制约其在焦化行业应用的关键因素之一。近年来,对VOCs的催化氧化已取得了很大进展,但鲜见催化氧化过程中催化剂的中毒机理和改善催化剂抗硫中毒性能的研究。基于此,笔者介绍了VOCs的催化燃烧机理、催化剂的硫中毒机理,论述了提高催化剂耐硫性的方法及耐硫性催化剂的表征技术,以期为设计和研发耐硫性能强的催化剂提供理论依据。
1 催化燃烧VOCs机理
催化剂中活性氧与催化剂的氧化还原性密切相关,提高催化剂的氧化还原能力可增强催化剂的耐硫性,因此提高活性氧浓度可以提高催化剂的耐硫中毒能力。了解VOCs的燃烧机理,确定与VOCs作用的氧物种类型对耐硫性催化剂的设计至关重要。虽然关于VOCs催化氧化作用研究较多,但由于污染物性质和反应条件不同,很难提供一个普遍和单一的反应机理,VOCs完全催化氧化的机理大致可分为三大类[7-9]:Marse-van-Krevelen (MvK)模型、Langmuir-Hinshelwood (L-H)模型、Eley-Rideal (E-R)模型。各模型的有效性很大程度上取决于催化剂的性能及VOCs的性质。
1.1 Mars-van-Krevelen机理
MvK模型认为吸附的VOCs与催化剂晶格氧发生反应,而不是与气相中的氧发生反应。该模型假设VOCs的氧化分为3步,如图1所示[10]。① VOCs 吸附在催化剂表面;② 吸附的VOCs与催化剂中氧发生反应,导致金属氧化物的还原;③ 还原的金属氧化物被进料中的气相氧再氧化,由于催化剂首先被还原,然后再被氧化,这种机制也被称为氧化还原机制。在稳定状态下,还原态的活性位中心将活性氧转移到氧化态的活性位上以提供VOCs反应所需氧,同时氧化态活性位将产生的电子传递给还原态活性位,使氧化还原循环往复进行[11]。
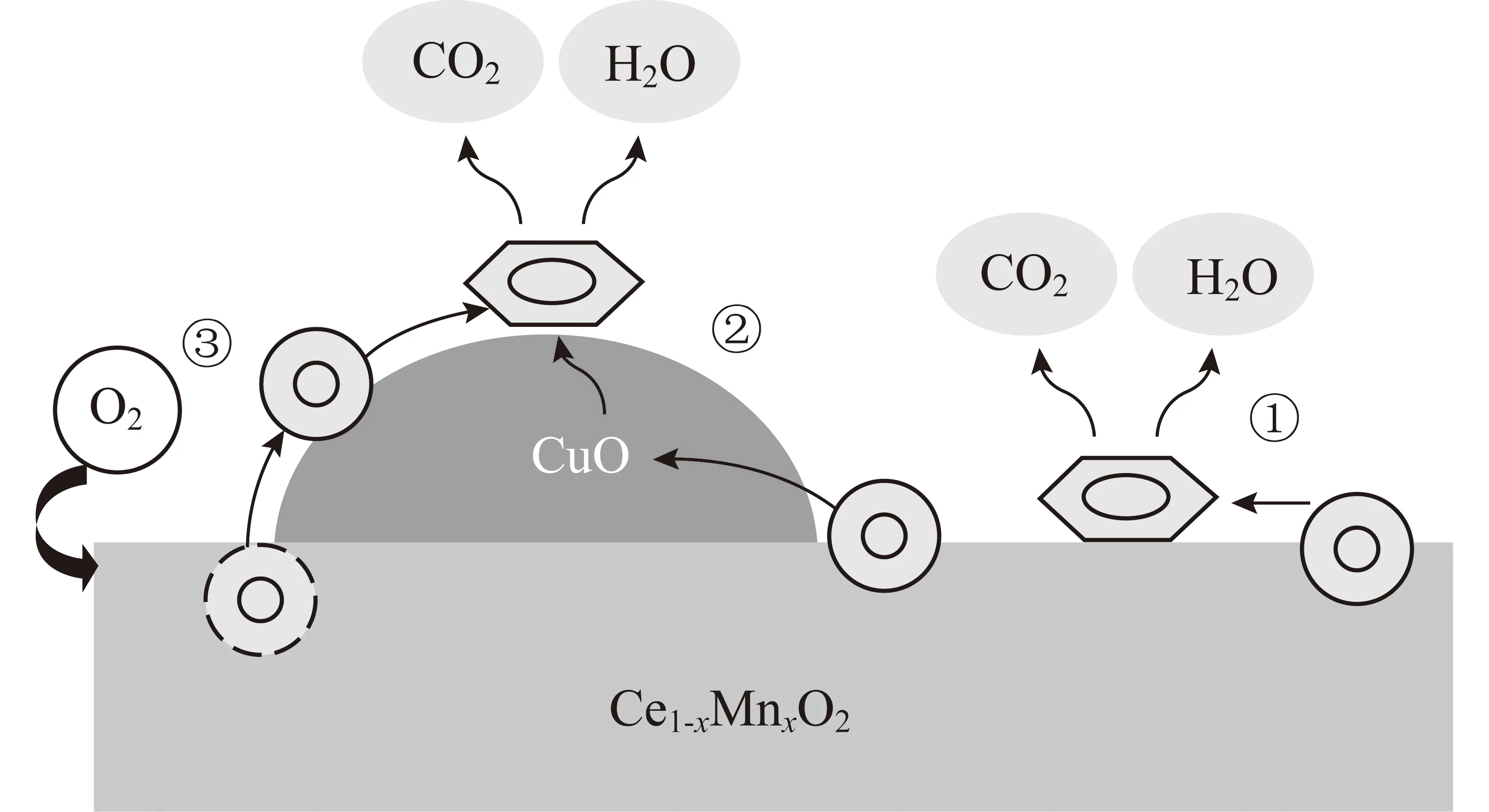
图1 苯在CuO/Ce1-xMnxO2上的氧化还原机理[10]Fig.1 Mechanism of benzene oxidation overCuO/Ce1-xMnxO2 catalysts[10]
MvK模型被广泛应用于烃类氧化反应的动力学模拟,特别是烃类氧化反应金属氧化物材料的动力学建模。脂肪烃类化合物中以甲烷为代表,大多数研究者认为催化剂中的晶格氧参与了反应,反应机理更符合MvK模型[12-14]。ZENG等[15]选取苯作为苯系物的代表,基于MvK机理,通过原位红外光谱试验得出苯在MnOx/TiO2材料上的具体氧化反应过程(图2)。
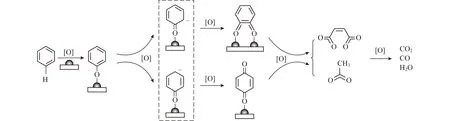
图2 苯在MnOx/TiO2催化氧化机理[15]Fig.2 Proposed mechanism for the catalytic oxidation of benzene over MnOx/TiO2 catalysts[15]
1.2 Langmuir-Hinshelwood机理
L-H机理假设反应发生在吸附的VOCs和吸附的氧之间[16-17]。因此,VOCs和氧物种必须吸附在催化剂表面,2种吸附分子的表面反应是该机理的控制步骤。L-H模型可根据VOCs分子和氧分子吸附位点的不同细分为单位点L-H模型和双位点L-H模型,单位点L-H模型中VOCs和氧吸附在相似类型的活性位点,双位点L-H模型中VOCs和氧吸附在2种不同类型的活性上。该模型的优点是不仅考虑了反应速率,还考虑了VOCs和氧气的吸附。HOSSEINI等[18]报道了甲苯和丙烯在Pd-Au/TiO2催化剂上的氧化反应遵循L-H模型,VOCs分子和氧分子在催化剂表面存在吸附竞争现象。BANU等[19]对甲基异丁基酮的氧化也得到了类似的结果。
1.3 Eleye-Rideal机理
根据Eleye-Rideal (E-R)原理,反应发生在被吸附的氧与气相中的反应物之间,控制步骤是吸附分子与气相分子之间的反应[20]。王放[21]发现甲苯催化燃烧反应过程,可用E-R机理解释短孔道Pd/Zr-Ce-SBA-15催化燃烧行为。另外,BANU等[22]发现环辛烷的Pt/γ-Al2O3催化氧化也遵循E-R机制。
每种机理的适用性在很大程度上取决于催化剂(活性金属和载体)与VOCs的性质。由于VOCs种类较多,催化反应过程比较复杂,机理分析仍处于初步探索阶段。但上述模型对金属氧化物和贵金属催化剂上挥发性有机物的氧化试验数据有一定的拟合性。活性氧类型和浓度与催化剂的耐硫性密切相关,因此,确定催化反应的机理,明析活性氧物种类型对耐硫性催化剂的设计至关重要。
2 催化剂硫中毒机理
中毒现象的本质是微量杂质与活性中心作用,形成了没有活性的物种。在催化燃烧过程中,对金属催化剂而言,H2S、H3P、SO2、COS、CO、Cl-等都是毒物,其中研究较多的是硫中毒[23]。催化剂硫中毒机理受多因素影响,如载体理化性质、VOCs组成、助剂类型等。以具有代表性的典型催化剂为例,分析其在硫化物存在时的中毒过程,总结催化剂硫中毒的影响因素及机理。
2.1 硫化物种类的影响
催化剂硫中毒机理受硫化物种类影响,研究发现甲烷燃烧PdO/Al2O3催化剂(甲烷是最难氧化的烃类化合物,VOCs燃烧可将甲烷作为模型化合物处理)在H2S和SO2中失活及再生机理不同。YU和SHAW[24]研究了PdO/Al2O3在过量氧气与H2S下的失活情况。根据研究结果和文献资料,提出了一个连续3个反应步骤的失活机理。在200 ℃时,动力学因素抑制了硫化物氧化为亚硫酸盐,因此PdS在低温下仍停留在催化剂表面。在较高温度(400 ℃)且有氧情况下,硫化物会氧化成亚硫酸盐,亚硫酸盐进一步与载体作用生成PdO活性组分。同时,YU和SHAW[24]通过红外光谱测量,解释了活性位点物理堵塞和比表面积减小的原因是Al2(SO4)3的形成。ORDEZ等[25]通过惰性气氛下600 ℃的高温脱附试验研究了PdO/Al2O3催化剂被SO2毒化后的失活和再生机理:

(1)
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
催化剂失活的根本原因在于SO2与催化剂活性组分和载体结合,产生亚硫酸盐或硫酸盐,使催化剂结构发生变化。HAMZEHLOUYAN等[26]采用程序升温脱附试验,研究了SO2、SO3和H2SO4处理后的Pt/Al2O3对硫的吸收和释放性能的影响,发现不同硫形态对催化剂硫中毒的影响程度从大到小依次为:H2SO4>SO3>SO2。
综上所述,硫化物种类会影响催化剂的耐硫性,原料气中硫的氧化程度越高,催化剂的耐硫性越差。因此,应尽可能减弱催化剂在催化燃烧VOCs过程中对低价硫化物的氧化程度,从而减弱硫化物与催化剂的相互作用来提高催化剂的耐硫性。
2.2 载体理化性质的影响
催化剂硫中毒机理受载体理化性质的影响,GRACIA等[27]研究发现随着SO2的加入,负载Pt的Al2O3和SiO2上的CO点火温度升高,与Pt/SiO2相比,Pt/Al2O3失活速度更缓慢,这种失活行为是由于Al2O3载体可作为硫的储槽,延缓活性组分硫中毒的发生。Pd基催化剂的失活机理及其在反应过程中载体的作用如图3所示。

图3 硫酸化和非硫酸化载体上PdO甲烷氧化活性的SO2抑制机理[28]Fig.3 Proposed mechanism for SO2 inhibition of PdO methaneoxidation activity for PdO on sulfating and non-sulfating support[28]
对于Pd基催化剂,PdO会促使SO2氧化为SO3,在可硫酸化载体上,PdO和载体都能吸附SO3,载体可作为SO3的储槽保护PdO,载体对SO3的吸附降低了催化剂的失活速率和失活程度。当原料气中停止加入SO2时,SO3会从载体溢流到PdO继续抑制甲烷氧化。而非硫酸化载体不能作为SO3储槽,PdO是SO3化学吸附的唯一目标,气相SO2氧化形成的SO3直接与PdO作用,导致催化剂失活速率较快。如果将SO2从原料气中去除,在约600 ℃以上时,SO3从PdO中解吸,催化剂的初始活性几乎完全恢复。然而,当原料气中再次加入SO2时,催化剂会迅速失活[28]。
载体是否可以硫酸化对催化剂的中毒机理及耐硫性有重要影响。可硫酸化载体可作为牺牲剂减缓硫化物对活性组分的毒化。但载体可硫酸化程度太强会大大破坏催化剂的结构,影响催化剂表面性质。因此选择合适的载体对耐硫性催化剂的设计尤为重要。
2.3 反应温度的影响
GREMMINGER等[29]发现硫酸盐的生成会导致Pd基催化剂的快速失活,如图4所示,温度高于550 ℃时,在富燃条件下催化剂活性恢复。WILBUR和EPLING[30]通过程序升温氧化、程序升温分解及程序升温还原,研究Pd-Pt/Al2O3催化剂受SO2毒化后的再生性,发现催化剂的活性不能经热分解得到恢复,但经程序升温还原至600 ℃后,只含Pd及0.9Pd-0.1Pt 组分的催化剂活性优于新鲜催化剂。
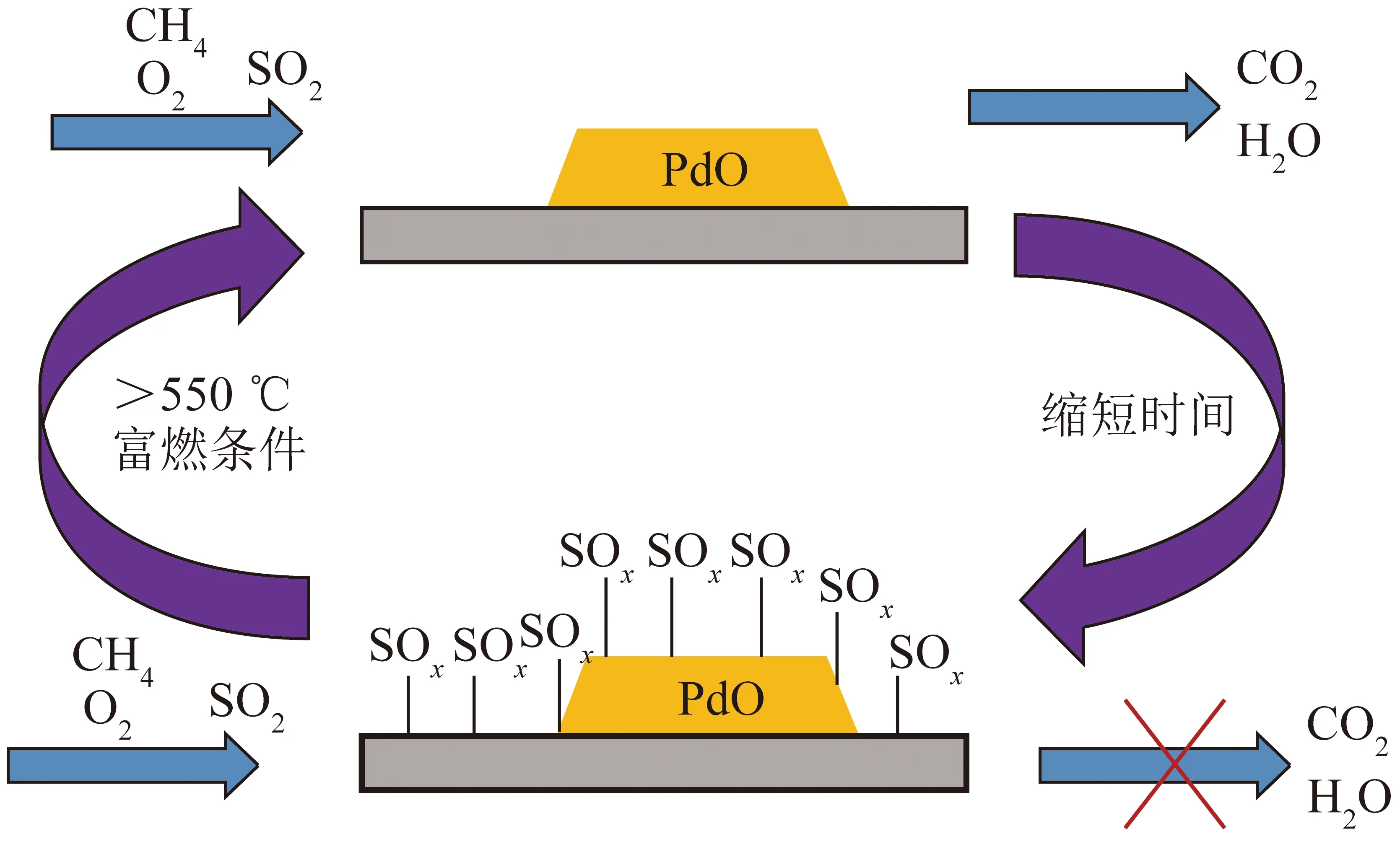
图4 Pd 基催化剂的中毒与再生[29]Fig.4 Poisoning and regeneration of Pd-based catalyst[29]
有些硫中毒后的催化剂通过热再生可在一定程度上恢复其活性,催化剂被毒化时生成的硫酸盐或亚硫酸盐的稳定性会影响其再生的难易程度。因此选择合适的催化剂体系和处理温度对催化剂的再生性能有很大影响。
3 催化剂耐硫性改善方法
VOCs燃烧过程中催化剂的氧化还原能力具有重要作用,不仅能提高催化剂活性,还可以增强催化剂的耐硫性,因此,可将提高催化剂氧化还原性能作为改善催化剂耐硫性的一种手段。研究多相催化反应过程,发现硫化物对催化剂的影响过程依次为:催化剂吸附硫化物→催化剂活性组分与硫化物作用→硫酸盐溢流→升高到一定温度的硫化物脱附。根据硫化物与催化剂间的作用过程调控催化剂的耐硫性具有可操作性,首先,通过调控催化剂对硫化物的吸附能力减少对硫化物的吸附;其次,添加助剂或使用可硫酸化载体,通过催化剂其他组分的竞争吸附来保护活性组分。此外,研究发现通过调控原料气体组成也可以加强催化剂的耐硫性能。
3.1 氧化还原性的增强
3.1.1形貌控制
催化剂形貌会影响活性氧数目及氧空位的形成。WANG等[31]通过水热氧化还原沉淀法制备了层状(LCMO)、棒状(RCMO)和粒状(GCMO)铜锰氧化物催化剂,发现层状LCMO催化剂对SO2表现出良好的耐受性。在SO2体积分数为50×10-6时,250 ℃甲苯转化率达100%,催化剂经1 000×10-6SO2预处理3 h后,在300 ℃粒状的GCMO几乎没有活性,而层状的LCMO催化剂对甲苯的转化率仍保持100%。主要原因是LCMO形成的具有大量缺陷的桥联式单斜四方相界面,可以抑制纳米颗粒的生长,使催化剂具有较小的颗粒尺寸和较大的比表面积,且混合相的界面结构可以诱导Cu2+-O2--Mn4+实体的形成,Mn—O和Cu—O键之间的相互作用使氧物种带更多的负电荷,这有利于氧物种的活化,产生更多的氧空位,从而提高了催化剂的氧化还原性。3种催化剂的CO或VOCs消耗率与氧空位的关系如图5所示,可知氧空位与VOCs的消耗率呈正相关,氧空位越多,VOCs的消耗率越大。赵伟荣和曾婉昀[32]制备了一种特殊形貌的球壳形催化剂Pt/CuO-CeO2,比表面积较大,能提供较多的活性氧位点,有利于VOCs在催化剂表面吸附,贵金属与稀土元素的结合大大提高了催化剂的耐硫性。

图5 3种催化剂的CO和VOCs消耗率与氧空位的关系[31]Fig.5 Correlation of the occurrence of oxygen vacancy sites tothe CO and VOCs consumption rates of three catalysts[31]
3.1.2相互作用协同
催化剂的氧化还原性能与组分间的相互作用密切相关。DENG等[33]采用水热法合成Ag-OMS-2,并测试其催化燃烧苯的性能。研究发现尺寸较小的K+与Ag+交换导致结晶尺寸明显减小,锰八面体缺陷增加,高含量Mn3+和Ag—O—Mn桥键的形成大幅增加活性氧,使K/Ag-OMS催化剂在苯燃烧方面表现出较高活性。CHEN等[34]采用氧化还原法合成了Ce-OMS,Mn、Ce氧化物间的相互作用大大增加了催化剂表面的氧空穴和晶格氧迁移能力,表现出较好的低温还原性能,苯、甲苯和邻二甲苯转化率为100%时温度都小于300 ℃。QU等[35]用Ag对Mn/SBA-15进行修饰,发现Ag/Mn为1/3时催化剂活性最好,MnO2、Mn2O3、Ag1.8Mn8O16的共存以及Ag与Mn的强相互作用表现出良好的协同作用,提高催化剂的氧化还原性,形成丰富的活性晶格氧,从而提高甲苯氧化的催化活性。因此,可利用催化剂组分间的相互作用来增强催化剂的耐硫性能。
3.1.3分散性提高
增大活性组分的分散性可以提高催化剂耐硫性。研究发现催化剂的氧化性能与活性组分的分散性密切相关,SHIN等[36]采用溶胶凝胶法和共沉淀法制备了催化剂Pd/SGCZA和Pd/PCZA,研究发现预硫化处理前共沉淀法制备的Pd/PCZA活性较好,T50(CO转化率为50%时的温度)为60 ℃,而Pd/SGCZA燃烧CO在75 ℃转化率为50%;硫化处理后,Pd/PCZA和Pd/SGCZA催化剂的T50分别升至152和123 ℃。催化剂经预硫化处理后,活性均有所降低,其原因是催化剂中毒导致活性组分分散性降低、活性氧物种减少、氧化还原能力下降。Pd基催化剂T50和Pd分散度的相关性如图6所示,可知催化剂的活性组分分散度越高,催化剂的氧化能力越强,耐硫性越好。CHEN等[37]报道了LaCoO3中加入铜可以降低活性组分的晶粒尺寸,从而间接提高了活性组分的分散性,Pd/La-Cu-Co-O表现出较好的抗硫中毒能力。

图6 Pd基催化剂T50和Pd分散度的相关性[36]Fig.6 Correlation between T50 and Pd dispersionof fresh and sulfur-aged catalysts[36]
催化剂的耐硫性与催化剂氧化还原性密切相关,通过提高催化剂的氧化还原能力来增强催化剂的耐硫性能。催化剂形貌、活性组分分散性、催化剂组分之间的相互作用对催化剂的氧化还原性能有很大影响。因此,可通过调控催化剂形貌、增大活性组分分散度和控制催化剂组分间的相互作用来增强催化剂的耐硫性。
3.2 硫化物吸附性能抑制
3.2.1活性组分调控
WILBURN和EPLING[38]通过DRIFTS和TPD研究了Pd-Pt/Al2O3中贵金属晶粒大小对SO2吸附、解吸以及硫酸盐物种形成的影响,如图7所示。可知粒径较小的催化剂易形成较多的硫酸铝种类,并在高温下分解。相比之下,颗粒尺寸较大的催化剂往往形成更多的低温分解和解析物质(SO2分子和亚硫酸盐物种)。进一步研究发现,双金属Pd-Pt/Al2O3催化剂的SO2吸脱附量随Pt含量的增加而减少,Pd/Pt为1/4时催化剂耐硫性最好[39]。可见,活性组分晶粒大小和比例会影响催化剂对硫化物的吸脱附能力。
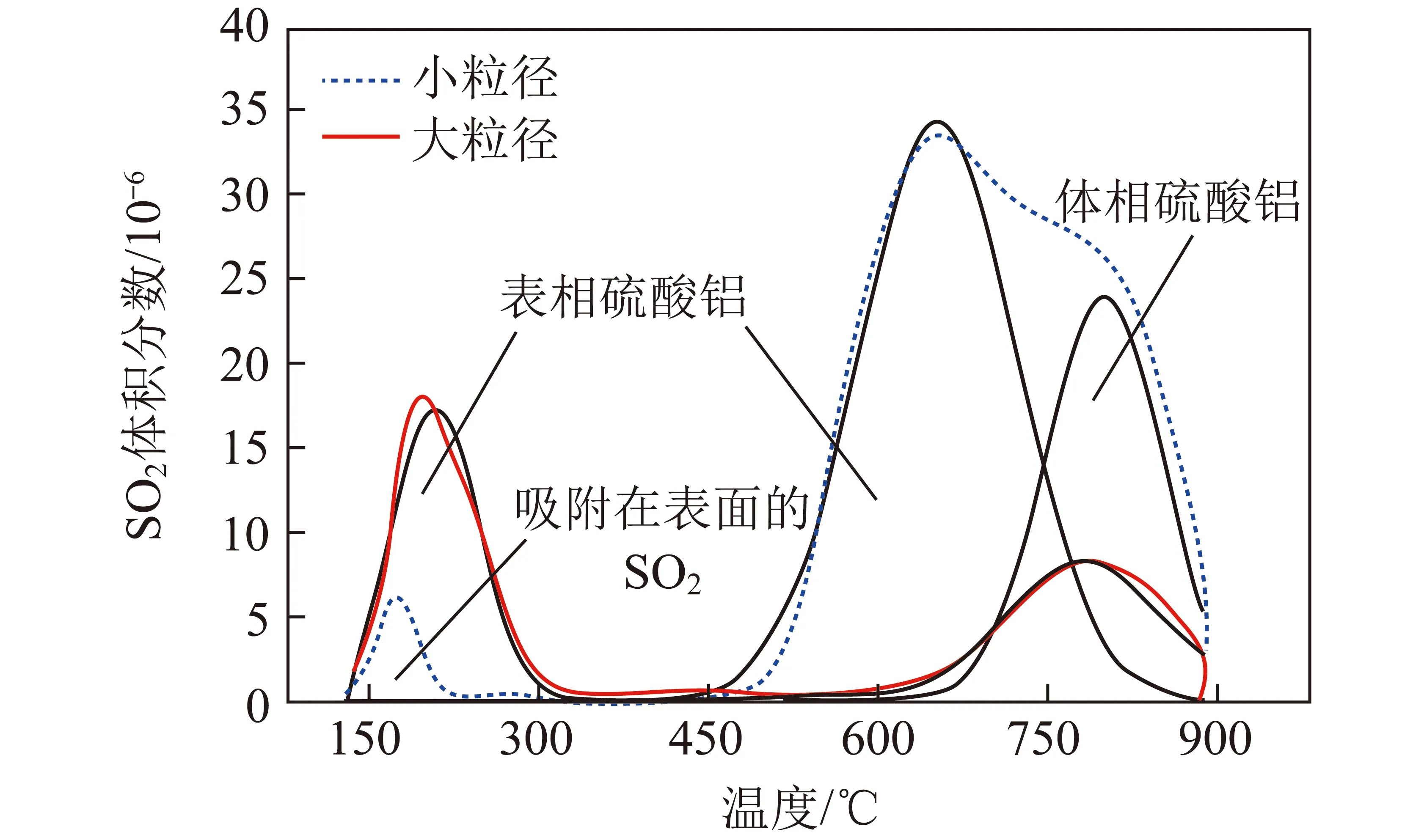
图7 2种Pd/Al2O3样品的SO2-TPD谱图[38]Fig.7 SO2-TPD spectra of two Pd/Al2O3 samples[38]
3.2.2酸碱性改变
ZHENG等[40]研究发现,在催化剂中加入Ni可提高还原性气氛下硫物种的解吸量,从而减少催化剂中硫物种的积累。刘建军[41]将Ni引入Cu/γ-Al2O3催化剂中,发现随着Ni含量增加,催化剂表面Lewis酸性降低,吸附SO2的能力减弱,可延缓催化剂硫中毒,同时也缩短了CO2分子在催化剂表面的停留时间,从而提高了甲烷催化燃烧效率。NIU等[42]研究发现在CuO/PdO/Al2O3中加入Sn有利于提高催化剂的耐硫性,这是由于SnO2为酸性氧化物,可以减少SO2在催化剂表面的吸附,从而减少硫酸盐物质在催化剂表面的生成。VENEZIA等[43]制备了纯二氧化硅SBA-15和HMS及相应的Ti4+改性介孔二氧化硅Pd基催化剂,发现TiO2的加入改善了SBA-15负载型催化剂的催化性能,提高了催化剂的耐硫性。最重要的是在后续无SO2运行中有利于催化剂的再生,而在Ti4+修饰的HMS载体上负载钯时,观察到相反的行为,活性较低,SO2的耐受性显著下降。基于结构和化学研究,认为这2种催化剂的不同之处在于HMS载体具有明显的结构和酸性,Ti4+更均匀地掺入HMS氧化硅结构中导致高酸性,催化剂呈强酸性会减少SOx的吸附,同时阻碍SOx与载体的优先反应,减弱TiO2的清除作用。
催化剂的抗硫中毒能力很大程度上取决于催化燃烧VOCs过程中对硫化物的吸脱附能力。具体表现为:粒径较小的催化剂易吸附硫化物形成较多的硫酸盐,并在较高温度下才能分解;而颗粒尺寸较大的催化剂往往会形成更多的低温分解和解析物质。但颗粒尺寸过大会减小活性组分分散度,影响催化剂活性和耐硫性。此外,可通过调控催化剂的酸碱性来提高其耐硫性。但催化剂酸性过强会抑制催化剂活性,酸性过弱会增加对硫化物的吸附能力。因此,合成具有适当粒径和酸碱性的催化剂对提高催化剂的耐硫性有重要意义。
3.3 活性组分的保护
3.3.1载体辅助
硫酸化载体可作为硫的储槽,减缓活性组分中毒。LIOTTA等[44]研究发现在有SO2存在的CH4燃烧过程中,催化剂Pd/Co3O4和Pd/CeO2均表现出一定的耐硫性。但CeO2比Co3O4更易与SO2作用,形成稳定的硫酸盐,对活性组分Pd的硫酸化有更好的保护作用,所以Pd/CeO2在硫的作用下表现出较强的耐硫性。MONAI等[45]系统研究了氧化铝负载的Pd@CexZr1-xO2纳米颗粒催化剂上甲烷氧化的SO2中毒情况,发现450 ℃时,SO2的存在会导致活性相PdO部分还原并在Pd位点上形成硫酸盐,约500 ℃时,硫酸盐会溢流到载体上与助剂作用,生成PdO使催化剂活性组分得到部分保护。
3.3.2助剂添加
刘新友等[46]合成了一种适合工业应用的耐硫性催化剂,向燃烧催化剂的涂层中加入CuO、MnO2、NiO、Fe2O3等成分,与活性组分Pd或Pt相比,助剂更易与硫化物结合生成硫酸盐,从而减弱催化剂中活性组分与含硫物质的相互作用。MA等[47]研究了Ba改性Pd/Al2O3甲烷燃烧催化剂,以硝酸钡为前驱体形成的Ba(NO2)2物种可抑制SO2和SO3从Al2(SO3)3和Al2(SO4)3向邻近活性组分PdO的溢出,提高催化剂的耐硫性。0.7% Ba-1% Pd/Al2O3,1% Pd/Al2O3和5% Pd/Al2O3催化剂在450 ℃时抗硫中毒性能如图8所示,可知0.7% Ba-1% Pd/Al2O3催化剂的耐硫性能接近高贵金属负载量5% Pd/Al2O3催化剂。
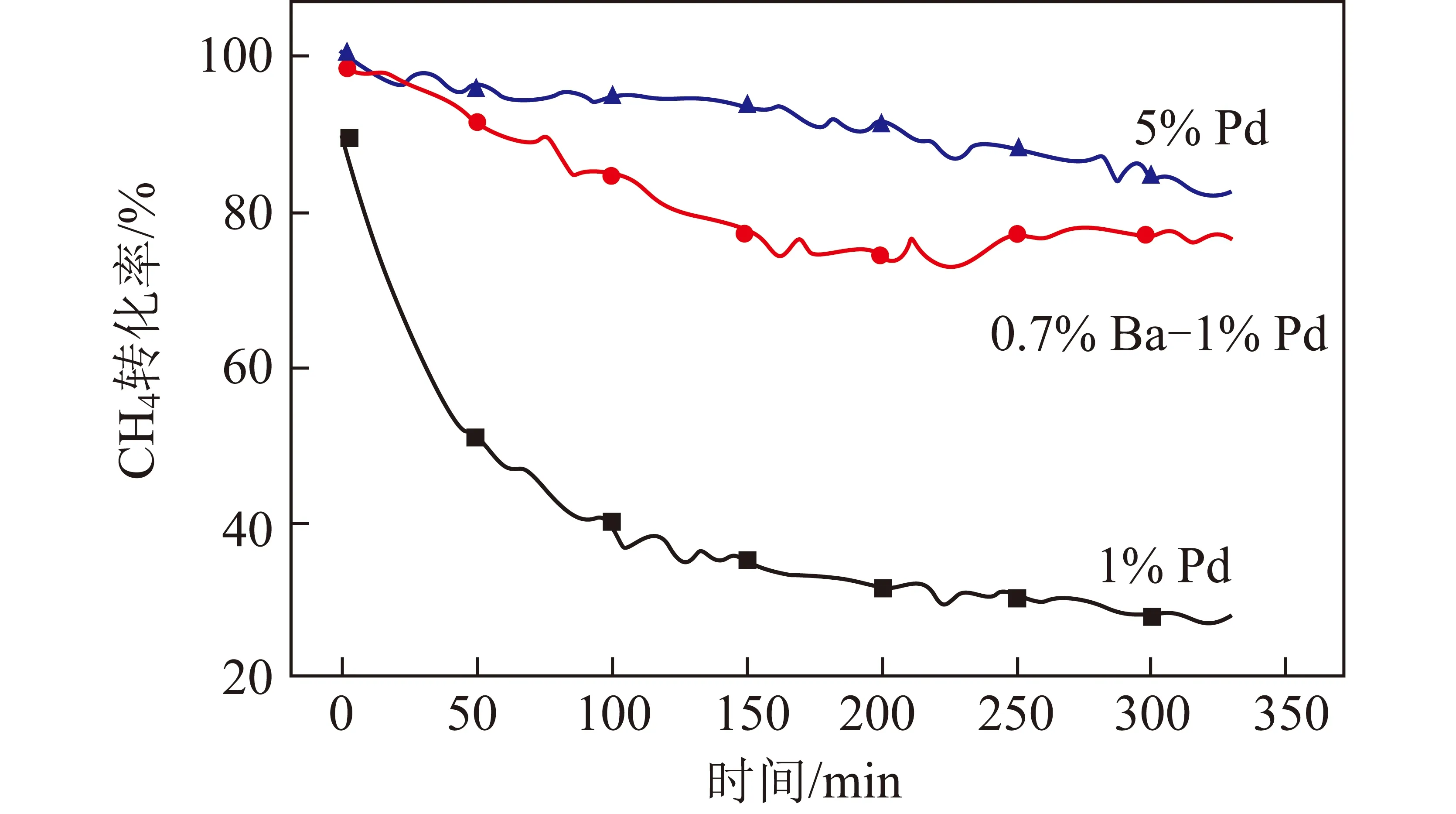
图8 3种催化剂在450 ℃反应温度下抗硫中毒性能[47]Fig.8 Performance of resistance to sulfurpoisoning of three catalysts at 450 ℃[47]
可硫酸化载体与助剂的引入均可改善催化剂的耐硫性,使活性组分得到保护,但在反应过程中,由于硫化物的存在会与催化剂组分作用生成硫酸盐,硫酸盐沉积在催化剂表面,导致催化剂整体结构遭到一定程度的破坏,比表面积下降,活性组分分散度降低,活性降低,缩短使用寿命。
3.4 水蒸气调控
一般情况下,水对催化剂的活性具有一定抑制作用,但也有研究报道水和硫化物在催化燃烧体系中共存时,可以阻碍硫酸根在催化剂表面的沉积,含有大量表面羟基的湿式硫酸盐催化剂比干式硫酸盐催化剂具有更高的表面酸性,从而促进催化活性[48]。刘宪云和高利平[49]考察了助剂MgO、K2O、ZrO2对催化剂耐硫性能的影响,认为助剂的加入均可提高催化剂的耐硫性,K2O效果尤为显著,T90(转化率为90%时的温度)仅比中毒前高了50 ℃,这是由于K2SO4熔点较高(1 069 ℃)难分解,但K2SO4易溶于水,水蒸气的存在可以促进硫脱附,有效提高催化剂的抗H2S中毒性能。因此,水蒸气调控可作为提高催化剂耐硫性的手段。但水蒸气调控有一定局限性,不适用于水热稳定性差的催化剂体系。
4 VOCs燃烧催化剂的表征手段
催化剂的耐硫性与催化剂组成、结构密切相关,通过一些化学表征技术了解催化剂的组分和物理化学结构,确定催化剂性质对催化剂耐硫性的影响十分重要。通过一些表征手段可以表征催化剂对SO2的吸脱附能力,确定VOCs在催化剂上的反应机理,明确活性氧物种,表征催化剂活性组分分散度。而SO2吸脱附能力、活性氧浓度和活性组分分散度等对催化剂的耐硫性有很大影响。CHEN等[37]通过SO2程序升温脱附(SO2-TPD)发现双金属催化剂Pd-Pt/Al2O3中SO2吸脱附量随着Pt含量的增加而减少;ZENG等[15]通过原位红外光谱试验(In situ-FTIR)阐述了苯在MnOx/TiO2上的催化氧化机理,证明了吸附氧在苯燃烧过程中起主要作用,为耐硫性催化剂的设计提供了理论依据;氧空穴对催化剂活性和耐硫性有很大影响;WANG等[31]通过电子顺磁共振光谱试验(EPR)发现氧空穴浓度与VOCs消耗率呈正相关;SHIN等[36]通过红外光谱(FTIR)研究了硫化处理后催化剂S-Pd/PCZA和S-Pd/SGCZA中的硫组分与催化剂组分的作用,发现硫在催化剂中以表面硫酸盐和体相硫酸盐2种形式存在;催化剂活性组分分散度与催化剂耐硫性能密切相关,SHIN等[36]采用CO化学吸附法计算Pd/Ce-Zr-Al-O中活性组分Pd的分散度。目前用于研究VOCs燃烧催化剂的主要表征技术见表1。

表1 耐硫性催化剂表征技术Table 1 Characterization technology of sulfur resistance of catalyst
由表1可知,XPS、FTIR、XRF均可表征催化剂中硫的存在形态进而解释催化剂的中毒机理;SO2-TPD和XRF可用来分析催化剂对硫化物的吸附能力;EPR、H2-TPR、O2-TPD可表征催化剂的氧化还原性能。其中一些技术在原位操作条件下可揭示中间产物生成及活性组分的催化氧化机理,获得一些有关催化剂硫中毒的重要机理信息。但这些表征手段仍存在一定局限性,如不能揭示催化剂在反应过程中的结构和性质变化及描述微观反应的动力学过程。目前,为了满足催化反应机理研究,在提升现代表征手段的同时,需不断强化计算模拟的辅助作用,表征+模拟的模式将成为未来研究催化剂耐硫性的趋势。
5 结语及展望
在京津冀、汾渭平原等地区,煤化工(焦化)废气是VOCs排放的重要工业源,催化燃烧技术因其低能耗、无副产污染物、适合大风量低浓度VOCs废气而具有良好的应用前景。针对催化燃烧法应用过程中催化剂硫中毒的问题,介绍了3种VOCs燃烧机理及3类催化剂中毒机理,归纳了提高催化剂耐硫性的手段,包括提高氧化还原性能、抑制硫化物吸附性能、保护活性组分和调控气体组成。这些手段均可在一定程度上改善催化剂的耐硫性。从而为解决困扰焦化行业的复杂VOCs治理问题提供理论支撑和试验基础,对山西省发展转型和清洁生产意义重大。
目前,耐硫催化剂研究面临以下问题:① 催化剂的硫中毒机理是根据其在反应前后的结构变化推测得出,不能揭示反应过程中催化剂失活的内在机理;② 在实际工况下,含硫成分复杂,对催化剂性能要求较高;③ 催化剂硫中毒机理的研究主要集中在贵金属催化剂,过渡金属催化剂的研究仍然很少; ④ 载体或助剂捕获硫化物后会改变催化剂结构,大大缩短催化剂的寿命。
VOCs燃烧耐硫催化剂未来研究重点在于:① 明确VOCs在催化剂表面的催化燃烧机理及硫化物对催化剂的毒化机制,调控催化剂材料表面抗性,以解决现有催化剂活性不足、耐硫性差等问题;② 针对现实工况硫成分复杂的问题,设计合成多活性位催化剂核心材料,调节和提高催化燃烧活性;③ 探究反应物的演变规律,为中间物的选择调变提供依据,丰富催化氧化反应理论体系;④ 在催化剂合成过程中优化操作条件,开发保留催化剂原结构条件下的新合成路线,提高催化剂耐硫性,为兼具高活性和耐硫性能的VOCs燃烧催化剂的开发提供设计思路。
猜你喜欢
金属热处理(2022年10期)2022-10-25
昆明理工大学学报(自然科学版)(2022年4期)2022-09-07
煤气与热力(2021年12期)2022-01-19
建材发展导向(2021年14期)2021-08-23
环境卫生工程(2021年3期)2021-07-21
当代水产(2021年3期)2021-07-20
食品安全导刊(2021年30期)2021-02-15
当代陕西(2020年23期)2021-01-07
当代化工(2019年3期)2019-12-12
火炸药学报(2014年1期)2014-03-20
