
基于16S rRNA高通量测序评价患病草鱼体表及肝脏细菌多样性
2022-03-20 12:20王晓磊付龙威刘学美张艳珍隋智海刘云国
水产科学 2022年2期
孙 坤,王晓磊,张 洁,付龙威,刘学美,张艳珍,隋智海,刘云国
( 1.临沂大学 生命科学学院,山东 临沂 276000; 2.临沂大学 教育学院,山东 临沂 276000; 3.新疆大学 生命科学与技术学院,新疆 乌鲁木齐 830046; 4.临沂市河东区农业技术推广站,山东 临沂 276003 )
草鱼(Ctenopharyngodonidellus)属鲤形目、雅罗鱼亚科、草鱼属,是一种典型的草食性淡水鱼类[1]。草鱼因具有生长快、易饲养、品质好等优良特性,已成为中国乃至世界上产量最大的淡水养殖鱼类之一[2]。随着养殖规模的不断扩大及养殖环境的逐步恶化,病原微生物感染频发,导致草鱼肌肉品质下降,口味变差,甚至引发大规模死亡,造成严重的经济损失[3]。草鱼养殖中常见的细菌感染性疾病有烂鳃病、细菌性败血症、肠炎病、赤皮病等,往往发病急、死亡率高[4-5]。烂鳃病主要是由柱状黄杆菌(Flavobacteriumcolumnare)引起,可导致草鱼烂鳃、体表溃疡等症状[6]。气单胞菌属(Aeromonas)可引发草鱼等淡水养殖鱼类的细菌性败血症、肠炎病[7-8]。目前,草鱼养殖中细菌感染的控制和预防主要是依赖抗生素和疫苗,如灭活嗜水气单胞菌疫苗等[9],但这些方法并不能完全避免病害暴发。
为实现鱼类细菌性疾病的精准防治,快速诊断并鉴定病原细菌、把握防治关键时机至关重要[10]。传统病原菌鉴定方法主要是基于分离—培养—分析的手段,如常规16S rRNA PCR鉴定、形态学分析、生理生化特性分析、耐药性分析等[11]。通过传统方法目前已成功分离并鉴定出很多致病菌,如李楠等[12]从濒死草鱼的内脏中分离并鉴定出3株病原细菌——霍乱弧菌(Vibriocholerae)、嗜水气单胞菌(A.hydrophila)和柱状黄杆菌,并测定其毒力和耐药性;胡钱东等[13]首次报道类志贺邻单胞菌(Plesiomonasshigeloid)是草鱼肌肉糜烂病的致病菌;张飘等[14]在贵州岑巩县某养殖场发病草鱼体内分离到1株维氏气单胞菌(A.veronii)GZQG2019。但由于有些病原微生物不能培养,或培养条件苛刻,或因丰度低难以分离,传统方法在病原微生物多样性检测中具有局限性,且分离培养法也比较耗时,不利于及时把握、控制传染的关键时机[15]。
16S rRNA高通量测序是一种快捷有效的微生物检测方法,可全面评价微生物多样性,能弥补传统方法的缺陷[16]。周本翔等[17]采用16S rRNA高通量测序技术发现,食物改变会显著影响草鱼肠道真菌物种组成。朱文根等[18]利用16S rRNA高通量测序技术研究发现,草鱼呼肠孤病毒感染可使草鱼肠道微生态发生紊乱。曾学琴等[19]用高通量测序法比较患乳房炎奶牛和正常奶牛的乳头擦拭子和牛奶中细菌的多样性,阐述了奶牛乳房炎关联微生物。
笔者选取2个不同养殖池中患病草鱼的溃烂皮肤和肝脏作为试验对象,基于Illumina MiSeqTM高通量测序技术和生物信息学分析研究患病草鱼皮肤和肝脏的细菌群落组成和多样性,推测引发此次疾病的优势病原细菌,为病害的精准控制提供研究基础。
1 材料与方法
1.1 样品来源与采集
2020年4月,山东省临沂市蒙阴县某草鱼养殖场暴发疾病,造成草鱼大量死亡。草鱼发病症状为:体表鳞片有脱落,臀鳍基部充血发红及其周围皮肤溃烂,肛门红肿,肝脏肿大、呈灰白色。从2个养殖池中分别随机选取3尾濒临死亡、体质量相当的患病草鱼,用75%乙醇擦拭鱼体表面,用无菌采样袋密封后,置于冰盒中运往实验室。
在无菌条件下,分别取同一养殖池的3尾患病草鱼的臀鳍基部周围溃烂皮肤及其肌肉组织,混匀后,分别编号为1号池—皮肤(1-1)和2号池—皮肤(2-1);沿着肛门处将其解剖,剪取同一养殖池的3尾草鱼的部分肝脏,混匀后,分别编号为1号池—肝脏(1-2)和2号池—肝脏(2-2),对上述4个样品立即进行DNA提取。
1.2 DNA提取与浓度测定
参照GeneJET Genomic DNA Purification Kit试剂盒说明书,对4份样品(约30 mg)进行研磨并提取基因组DNA,采用1%琼脂糖凝胶电泳检测DNA的完整性,用Nanodrop仪器测定DNA的浓度,以确保所提取DNA质量满足后续扩增要求。提取的基因组DNA保存在-20 ℃。
1.3 16S rRNA基因扩增和测序
16S rRNA基因扩增和测序均由生工生物工程(上海)股份有限公司完成。针对16S rRNA基因的V4区域进行PCR扩增,设计带Barcode的特异引物515F:5′-GTGCCAGCMGCCGCGGTAA-3′;806R:5′-GGACTACHVGGGTWTCTAAT-3′。PCR扩增采用Phusion Flash high-fidelity PCR Master mix试剂盒,反应条件为:94 ℃预变性30 s;94 ℃变性30 s,50 ℃退火55 s,72 ℃延伸30 s,共30个循环;72 ℃延伸15 min。琼脂糖凝胶电泳回收PCR产物,利用Qubit 3.0 DNA检测试剂盒测定回收的DNA浓度,以便按照1∶1等量混合后测序。等量混合时,每个样品DNA量取10 ng,最后使用Illumina MiseqTM进行上机测序。
1.4 数据处理与分析
IlluminaMiseqTM测序获得原始序列(Raw data),利用Cutadapt、PEAR和Prinseq软件去除引物接头、拼接和去除低质量序列,得到有效序列(Clean data)。对有效序列完成运算分类单位(OTU)聚类、α多样性、β多样性和群落结构组成等分析[17-18]。
2 结果与分析
2.1 测序数据和聚类分析
提取2个不同养殖池的患病草鱼的臀鳍基部溃烂皮肤组织和肝脏DNA,扩增16S rRNA基因的V4区,基于Illumina MiSeqTM高通量测序检测体表和肝脏的细菌多样性,将原始数据过滤并统计分析,发现4个样本共得到原始序列186 963条,通过去除嵌合体及非特异性扩增序列后,得到有效序列共183 574条,平均测序读长为251.37 bp,每个样品有效序列占原始序列的比例均在96%以上(表1)。

表1 16S rRNA高通量测序数据统计Tab.1 Statistics of the high-throughput 16S rRNA sequencing data
在97%相似度的标准下,对测序序列进行聚类分析,发现1号池—皮肤和肝脏(1-1和1-2)的运算分类单元数分别为611个和875个,2号池—皮肤和肝脏(2-1和2-2)的运算分类单元数分别为376个和644个,其中4组样本所共有的运算分类单元数为87个,分别占总运算分类单元数的14.32%、9.94%、23.14%和13.51%;4组样品所特有的运算分类单元数目分别为342、584、209和384个(图1)。
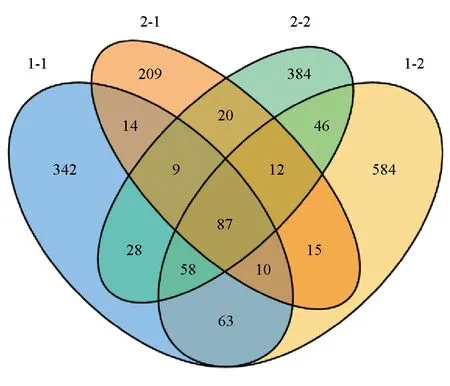
图1 运算分类单元分布韦恩图Fig.1 Venn diagram showing the distribution of OTU
2.2 稀释曲线
稀释曲线可评价测序量是否足以覆盖所有物种类群,并间接反映物种的丰富程度。由稀释曲线(图2)可见,随机抽取测序条数在30 000条以下时,随着测序条数增加,4个样品的运算分类单元数急剧上升;在30 000~50 000条,随着测序条数增加,4个样品虽然会有新的运算分类单元出现,但是曲线趋于平缓,表明该样品测序量合理,可真实反映出细菌群落结构和多样性。

图2 稀释曲线Fig.2 The dilution curve of samples
2.3 微生物多样性
2.3.1 α多样性
试验中所得到的患病草鱼皮肤和肝脏样品α多样性指数见表2。每个样品的覆盖率均达到99.4%以上,表明测序结果可靠,样品中的序列几乎全被检测出且达到饱和状态,可充分反映样品的细菌多样性。通过比较这4个样品各项的α多样性指数,发现1号池—肝脏(1-2)的Chao1指数和ACE指数最大,分别高达1375.109和1208.399,表明1号池的患病草鱼肝脏样品菌群丰度最大;1号池—肝脏(1-2)的Shannon指数最大,为3.298,Simpson指数最小,为0.24369,表明1号池的患病草鱼肝脏样品细菌群落多样性最高。此外,2个养殖池的患病草鱼微生物丰度和多样性肝脏(1-2和2-2)均比皮肤(1-1和2-1)高。
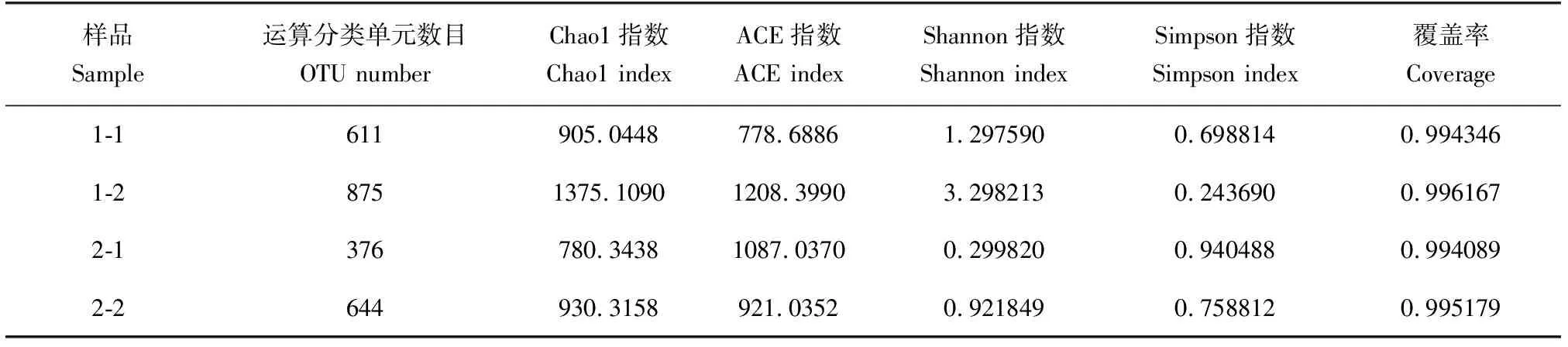
表2 样品细菌丰富度和多样性指数Tab.2 The bacterial abundance and diversity index of samples
2.3.2 β多样性
样品间微生物群落结构差异可通过β多样性来分析。基于运算分类单元进行主坐标分析得到不同样品的β多样性的主坐标分析(PCoA)(图3):第1主坐标和第2主坐标的贡献率分别为91%和9%;1号池—皮肤(1-1)、2号池—皮肤(2-1)和2号池—肝脏(2-2)这3个样品聚集在一起,距离较近,

图3 基于样品运算分类单元的主坐标分析Fig.3 PCoA analysis of sample OTUS
这表明这3个样品菌群结构较为相似;而1号池—肝脏样品(1-2)单独分布,距离其他3个样品距离较远,说明该样品与另外3个样品的菌群结构差异较大。
根据对不同样品进行聚类分析研究其微生物群落多样性差异,利用非加权组平均法(UPGMA)算法构建树状结构,再根据样本间Unifrac距离矩
阵绘制热图(图4,颜色块代表距离值,颜色越红表示样本间距离越近,相似度越高,越蓝则距离越远):1号池—皮肤(1-1)、2号池—皮肤(2-1)及2号池—肝脏(2-2)这3个样品距离很近;1号池—肝脏(1-2)则与其他3个样品距离较远。
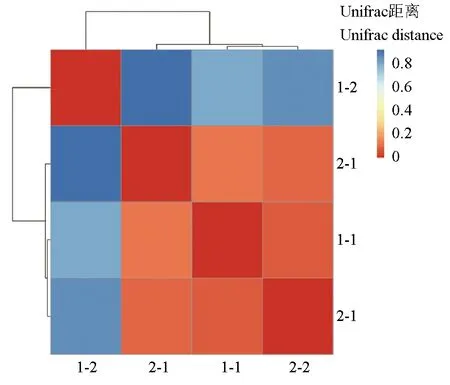
图4 基于Unifrac距离的热图Fig.4 Heatmap based on Unifrac distance
2.3.3 细菌群落结构
在门水平上,4组样品占优势的主要以拟杆菌门和变形杆菌门为主,但门的丰度有所差别,1号池—皮肤(1-1)、2号池—皮肤(2-1)和2号池—肝脏(2-2)均以拟杆菌门丰度最高,分别占84.78%、97.18%和87.37%,而1号池—肝脏(1-2)以变形杆菌门丰度最高,占80.02%(图5)。

图5 门水平上各样品中细菌群落的相对丰度和组成Fig.5 Relative abundance and composition of bacteria from different samples at the level of phylum
在属水平上,4个样品主要菌群丰度差异较大(图6)。1号池—皮肤(1-1)主要以黄杆菌属(Flavobacterium,83.38%)、鞘氨醇单胞菌属(Sphingomonas,1.63%)为主;1号池—肝脏(1-2)主要以希瓦氏菌属(Shewanella,49.39%)、鞘氨醇单胞菌属(4.39%)、不粘柄菌属(Asticcacaulis,2.07%)、黄杆菌属(2.05%)为主;2号池—皮肤(2-1)主要以黄杆菌属(96.93%)为主;2号池—肝脏(2-2)主要以黄杆菌属(86.88%)、耶尔森氏菌属(Yersinia,4.66%)为主。
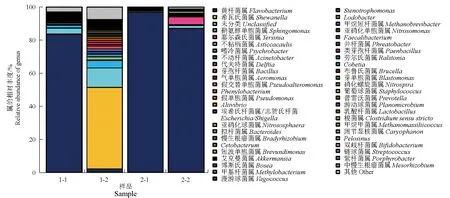
图6 属水平上各样品中细菌群落的相对丰度和组成Fig.6 Relative abundance and composition of bacteria from different samples at the level of genus
3 讨 论
草鱼作为我国四大家鱼之一,是重要的淡水养殖经济鱼。随着养殖规模扩大及部分养殖环境恶化,草鱼病害频发导致死亡,造成严重的经济损失[20]。因此,探索并诊断诱发病害的主要病原菌,了解其生物学特性,可为鱼类健康养殖及早期病害防治提供科学依据。
3.1 16S rRNA高通量测序技术的应用
传统方法是基于细菌分离培养法,通过形态特征、生理生化反应、16S rRNA基因序列测定与系统发育树分析鉴定鱼类病害的主要病原细菌,如邹升等[21]从湖南环洞庭湖区精养鱼池患病草鱼肠道中分离出气单胞菌、类志贺邻单胞菌和格氏乳球菌(Lactococusgarvieae)。但是这种方法比较耗时,且有些细菌不可培养,极大地限制了对病原细菌的认识和了解。
高通量测序技术是新型病原检测手段,具有快速、精准、阳性率高等特点,非常适合病原体诊断[22]。如任迪等[23]利用宏基因组高通量测序法显著提高了脓毒症患者血样本中的病原体检出率,缩短了检出时间;涂飞云等[24]利用16S rRNA高通量测序技术分析豪猪脓肿脓汁细菌多样性,为病原分离鉴定提供优势菌群的参考。
3.2 患病草鱼皮肤和肝脏的细菌多样性分析
本试验针对山东省临沂市蒙阴县某草鱼养殖场2个暴发病害的不同草鱼养殖池(1号池和2号池),利用16S rRNA高通量测序法分析患病草鱼的溃烂皮肤组织和肝脏的细菌多样性,快速诊断引发病害的病原细菌,为后期病害防治提供科学依据和参考。
16S rRNA高通量测序结果显示,总体上,1号池草鱼的皮肤和肝脏的运算分类单元数均比2号池草鱼皮肤和肝脏的运算分类单元数多,说明1号池内细菌感染情况较2号池复杂;α多样性分析结果显示,同一池内病鱼肝脏中微生物丰度和多样性较皮肤高,其中1号池—肝脏的菌群丰度和多样性最高,同样提示1号池内细菌感染情况较2号池复杂;β多样性分析结果显示,1号池—皮肤、2号池—皮肤和2号池—肝脏细菌群落结构相似,但1号池—肝脏细菌群落结构与其他3个样品存在差异,这表明1号池在与2号池暴发相同病原菌感染的同时,其内部可能并发了其他病原菌的感染,需要着重加以防治以免造成更大损失。曾有研究者发现,水温高、有机物含量高等会加强黄杆菌的黏附和侵染能力[25],因此推测2个养殖池的菌群结构差异可能与水温、水质等因素有关。
3.3 患病草鱼皮肤和肝脏的菌群结构分析
2个养殖池的草鱼皮肤菌群结构在门水平上均以拟杆菌门为主,在属水平上以黄杆菌属为主,这与正常鱼类体表细菌群落以变形杆菌门为主有所不同[26-29]。2号池—肝脏菌群结构与皮肤的相似,主要以拟杆菌门、黄杆菌属为主。黄杆菌属是一种机会病原菌,普遍存在于养殖水环境或泥土中,在健康鱼的外表能形成生物膜,抵抗黏膜免疫防御,进而感染多种野生和养殖淡水鱼种,导致其出现烂鳃、体表溃疡等症状[30-31]。
1号池—肝脏菌群结构中,同样发现有黄杆菌属(2.05%),但以变形菌门、希瓦氏菌属为主,与1号池—皮肤菌群结构不同,推测1号池在暴发黄杆菌属病原菌感染的同时,并发希瓦氏菌属感染。希瓦氏菌属也是一种机会病原菌,广泛存在于水环境中,可与黄杆菌属共同感染鱼类[32-33]。之所以1号池病鱼的肝脏菌群结构与皮肤存在差异,可能与某些病原菌的组织特异性相关,如黄杆菌属更趋向侵染皮肤黏膜系统[34];也可能与部分病原菌如希瓦氏菌等会对其他细菌存在拮抗作用有关[35]。此外,黄杆菌属和希瓦氏菌属目前已在虹鳟(Oncorhynchusmykiss)、斜带石斑鱼(Epinepheluscoioides)、大西洋鲑(Salmosalar)、河鲈(Percafluviatilis)、银大马哈鱼(Oncorhynchuskisutch)等中被分离鉴定,均能引发相应的鱼类疾病[36-38]。因此,我们推测这次病害极有可能是由黄杆菌属和希瓦氏菌属联合引发的,这需要进一步开展病原分离、鉴定以及接种感染试验,确定引发此次病害的病原细菌。
4 结 论
通过16S rRNA高通量测序法比较分析了临沂市某草鱼养殖场的2个不同的养殖池中的患病草鱼溃烂皮肤和肝脏的菌群多样性和结构组成,结果显示,1号池草鱼感染情况较2号池复杂;患病草鱼溃烂皮肤和肝脏菌群结构主要以拟杆菌门和变形杆菌门为优势菌门,以黄杆菌属和希瓦氏菌属为优势菌属,推测其是引发本次草鱼病害的优势病原细菌。该试验结果可为针对性防治该养殖场草鱼病害提供科学依据和参考。
猜你喜欢
今日农业(2022年14期)2022-09-15
当代水产(2022年5期)2022-06-05
卫星应用(2022年3期)2022-05-23
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年11期)2022-05-13
当代水产(2022年3期)2022-04-26
当代水产(2022年2期)2022-04-26
中国新通信(2022年4期)2022-04-23
大众健康(2021年8期)2021-08-04
广东第二课堂·小学(2018年9期)2018-10-24
