
线粒体基因和核基因揭示自然屏障和第四纪更新世气候振荡影响双斑乙蠊谱系地理格局
2022-03-17 06:57:22牛力康端木浩楠王宗庆车艳丽
昆虫学报 2022年2期
陈 蓉, 牛力康, 端木浩楠, 王宗庆, 车艳丽
(西南大学植物保护学院昆虫研究所, 重庆 400715)
1 INTRODUCTION
Sigmellabiguttata, belonging to Blattellinae, is distributed mainly in southwestern, southern and eastern China (Bey-Bienko, 1954; Li, 2018). Based on morphological characteristics and molecular data (COIand 28S rRNA), Li (2018) discovered that the morphological species,S.biguttata, was divided into several molecular operational taxonomic units ( MOTUs), which exhibited some minor morphological variations among different geographic populations, and then speculated that the significant genetic differentiation might exist in different populations.
Recent phylogeographic study (Liuetal., 2018) showed that geographical barriers and Quaternary climatic oscillations were important factors causing the genetic differentiation and geographic distribution pattern of the East Asia cicada,Hyalessamaculaticollis. As the geographical barriers usually reduced gene flow and contributed to population differentiation of the Chinese hwamei,Leucodioptroncanorumcanorum(Lietal., 2009), and organisms survived in refuge during glacial periods, then expanded after the ice age, and eventually formed their current distribution, such as the moth,Apocheimacinerarius(Liuetal., 2016). Medium body size, humus feeding and low dispersal ability ofS.biguttatalead itself to being as a model organism for phytogeographic study.
Some studies have shown that the mitochondrial genesCOI(Pramualetal., 2005),COII(Zhouetal., 2012) andND1 (Chenetal., 2019) were excellent gene makers to analyze the genetic differentiation and population expansion, and nuclear gene (ITS) could make up for the shortcoming of mitochondrial gene and accurately discover the population migration history (Bensassonetal., 2001). Therefore, mitochondrial and nuclear genes are combined to reflect the genetic differentiation and infer the causes of the geographical distribution pattern more comprehensively (Liuetal., 2018).
In this study, the genetic diversity, genetic differentiation and phylogeographic structure of 284 samples from 19 geographic populations ofS.biguttatawere analyzed based on three mitochondrial genes (COI,COIIandND1) and one nuclear gene (ITS) to infer the time of population divergence, to reveal the population historic dynamics, and to explore the cause of the geographic distribution of this species.
2 MATERIALS AND METHODS
2.1 Sample collection, DNA extraction and PCR amplification
The 284 samples of 19 geographic populations ofS.biguttataand 2 samples of the closest relatives (SigmelladigitalisandSigmellanormalis) used in this study were collected during 2013 and 2020 (Table 1). The specimens were immersed in 100% ethyl alcohol and then stored at -20℃. The voucher specimens were deposited in the Institute of Entomology, Southwest University (Chongqing).
Complete genomic DNA was extracted from leg muscle ofS.biguttatausing the protocol of the Small Tissue DNA Extraction Kit following the manufacturers’ instructions. Three mitochondrial genes (COI,COII, andND1) and one nuclear gene (ITS) were amplified and sequenced. All primers were designed using Primer Premier v.5.0 (Lalitha, 2000) and synthesized by Tsingke Biological Technology (Beijing) shown in Table 2. PCR was conducted in 25 μL reaction mixture including 22 μL of T3 Mix, 1 μL of DNA template, and 1 μL of each primer (10 μmol/L). Reactions were conducted in a SimpliAmp Thermal Cycler with an initial denaturation at 98℃ for 2 min followed by 35 cycles of denaturation at 98℃ for 10 s, annealing at 49-56℃ (depending on the primer pairs) for 10 s, and elongation at 72℃ for 50 s, with a final extension step at 72℃ for 10 min. Amplifications were tested using 1.0% agarose gel electrophoresis and sent to Tsingke Biological Technology (Beijing) for sequencing.
2.2 Data analyses
Nucleotide sequences ofCOI,COII,ND1, andITSof geographic populations ofS.biguttatawere aligned using the online MAFFT server (https:∥mafft.cbrc.jp/alignment/server/) under the G-INS-i (COI,COII,ND1) or Q-INS-i (ITS) algorithm. Then the manual adjustment was performed in MEGA v.7.0 (Kumaretal., 2016). Population genetic parameters including the number of haplotypes (Nh), haplotype diversity (Hd) and nucleotide diversity (π) for each population and overall populations were calculated using DnaSP v.5.0 (Librado and Rozas, 2009).
Spatial analysis of molecular variance (SAMOVA) was completed in SAMOVA v.1.0 (Dupanloupetal., 2002) based on combined dataset (COI+COII+ND1+ITS).Kvalues (number of groups) were set from 2 to 12 and each independent simulated annealing process was performed with 100 replicates. Analysis of molecular variance (AMOVA) was completed in Arlequin v.3.5 (Excoffier and Lischer, 2010).Fstwas estimated in Arlequin v.3.5 to quantify the differentiation between pairwise populations and pairwise genetic groups, with statistical significance tested by 1 000 non-parametric permutations at the 5% significance level. Gene flow (Nm) can be calculated according toFst,Nm=(1-Fst)/4Fst(Wright, 1949).
2.3 Phylogenetic constructions and haplotype networks
WithS.digitalisandS.normalisdesignated as outgroups, phylogenetic analyses were performed based on combined dataset (COI+COII+ND1+ITS) using maximum likelihood (ML) (Stamatakisetal., 2008) and Bayesian inference (BI) methods (Ronquistetal., 2012). The best nucleotide substitution model (COI: TIM+G;COII: TrN+I;ND1: K81uf+I+G;ITS: JC+I) was selected by PartitionFinder v.1.1 (Lanfearetal., 2012). Haplotype networks were constructed using median joining network algorithm in PopART v.1.7 (Leigh and Bryant, 2015).

Table 1 Sampling information, and genetic parameters based on combined dataset (COI+COII+ND1+ITS)of Sigmella biguttata geographic populations and sampling information of outgroup species

Table 2 PCR primers used in this study
2.4 Population historic dynamics analysis
Neutrality test and mismatch distribution analysis were used to detect the population historic dynamics in Arlequin v.3.5 based on combined dataset (COI+COII+ND1+ITS). Tajima’sD(Tajima, 1989) and Fu’sFs(Fu, 1997) values were used to detect the expansion and equilibrium of population. Significant negative values represent population expansion (Fu, 1997). An observed unimodal distribution indicates population expansion, while an observed multimodal represents an equilibrium population (Rogers and Harpending, 1992). In addition, the sum of squared deviations (SSD) and Harpending’s raggedness index (r) values can be used to test the goodness of fit of the observed and expected distributions. The expansion time (t) can be calculated using the formulat=τ/2 μk (k=3 409, the length of the sequence; μ=0.0177, the substitution rate ofCOIgene) (Rogers and Harpending, 1992).
2.5 Divergence time estimation
The timing of divergence was estimated using the software BEAST v.1.10.4 (Suchardetal., 2018) based on combined dataset (COI+COII+ND1+ITS). Outgroups and the nucleotide substitution model of each gene followed the phylogenetic analysis. Birth-Death Process was used as the tree prior model, and strict clock as the molecular clock model (Brown and Yang, 2011). The substitution rate ofCOIgene was 0.0177 per site per million years (Papadopoulouetal., 2010). The Markov Chain Monte Carlo (MCMC) chains were conducted for 100 million generations with a sample frequency of 1 000. Convergence was checked by the effective sample sizes (ESSs)larger than 200 in Tracer v.1.7 (Rambautetal., 2018).
3 RESULTS
3.1 Genetic diversity and population genetic structure of S. biguttata
The final combined dataset (COI+COII+ND1+ITS) (the same below) included 284 sequences with the length of 3 409 bp in all (COI: 658 bp;COII: 728 bp;ND1: 945 bp;ITS: 1 079 bp). All new sequences have been deposited in GenBank with accession numbers MZ818048 to MZ818333 forCOI, MZ825562 to MZ825847 forCOII, MZ825848 to MZ826133 forND1 and MZ810639 to MZ810922 forITS. Sequence analysis revealed a total of 610 variation sites, 201 polymorphic sites, 169 singleton sites and 139 haplotypes. The total populationHdandπwere 0.9880 and 0.0278, respectively, indicating high genetic diversity ofS.biguttata. In each geographic population (Table 1), the genetic diversity of populations HiN and FJWYS was distinctly higher than that of the others.
SAMOVA analysis based on the combined dataset showed thatFctvalues (the genetic differentiation among groups) gradually rose fromK=2 to 8 and decreased fromK=9 to 12.Fctreached the largest value (Fct=0.77) whenK=8. Eight groups were recovered as the optimal partition of populations (more details in Table 3). Most groups had higher genetic diversity (Hd>0.5,π>0.005), and Group 4 and Group 6 had low nucleotide diversity (π<0.005).
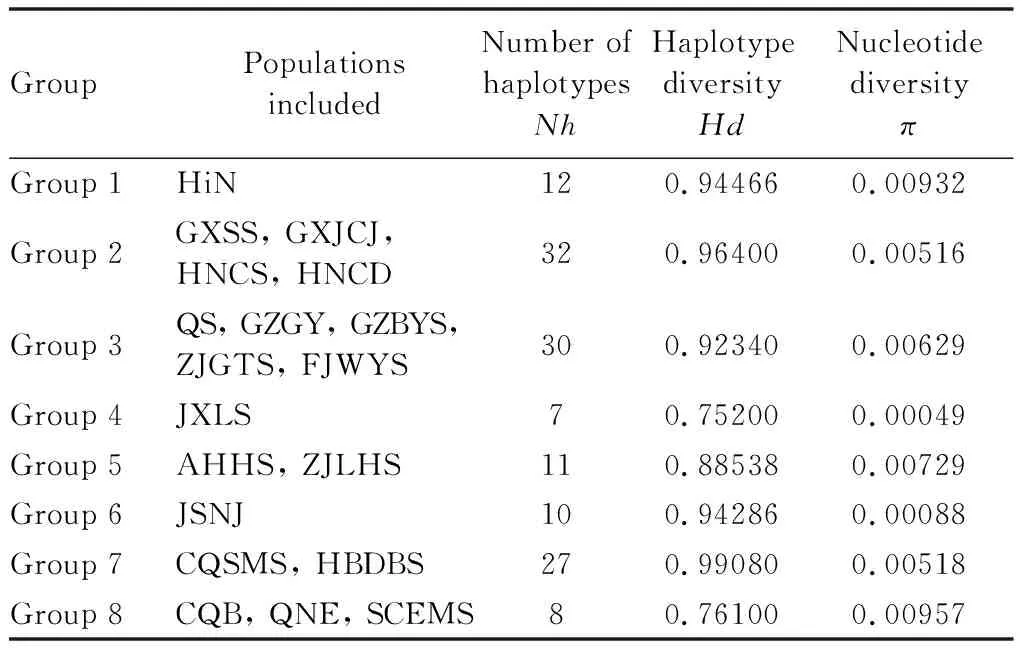
Table 3 Genetic diversity of each group of Sigmellabiguttata populations based on combined dataset(COI+COII+ND1+ITS)
AMOVA analysis (Table 4) showed significant differentiation among groups (77.09% of the variation) compared to the level among populations (16.05%) or within populations (6.86%). In addition, the pairwiseFstvalues among populations and among groups were 0.43083-0.99746 (>0.25) and 0.66746-0.97967 (>0.25), respectively (Fig. 1: A, B). The maximum gene flow (Nm) among populations and among groups were 0.33 and 0.12 (<1), respectively (Fig. 1: C, D). All the above results indicate that there is significant genetic differentiation inS.biguttata.

Table 4 AMOVA analysis results of Sigmella biguttata populations based on combined dataset(COI+COII+ND1+ITS)

Fig. 1 Fst and Nm values among populations and groups of Sigmella biguttata based on combined dataset (COI+COII+ND1+ITS)A: Fst value of population; B: Fst value of group; C: Nm value of population; D: Nm value of group.
3.2 Phylogenetic analyses of S. biguttata
Our phylogenetic analyses based on combined dataset (COI+COII+ND1+ITS) (Fig. 2: A) showed that the maximum likelihood and Bayesian inference trees yielded almost identical topologies with generally high support values. All populations of eight groups were clustered together to be three lineages, which was consistent with the result of SAMOVA analysis. HiN population (lineage I) was discovered to be the sister of the mainland populations (lineages II, III). The same geographic populations were clustered together to form one single branch, suggesting obvious phylogeographic structure.
The haplotype network based on combined dataset (COI+COII+ND1+ITS) (Fig. 2: B) showed that the population structure was similar to that retrieved in the phylogenetic analyses, with eight groups distinguished. There was no shared haplotype among different geographic populations, and haplotypes from the same population were clustered together. Populations in lineages I and II, and lineages II and III are both connected by as many as about 60 mutation steps. The existence of many mutation steps among haplotypes and no shared haplotypes among different geographic populations indicated that significant genetic differentiation exists among populations ofS.biguttata, which was consistent with the results obtained by AMOVA analysis and genetic differentiation index.
3.3 Population historic dynamics of S. biguttata
Neutrality test (Table 5) showed that Tajima’sDand Fu’sFsvalues of Group 1, Group 3, Group 5 and Group 8 were all positive, indicating that these populations are stable without expansion, which was consistent with the multi-peak results of mismatch distribution (Fig. 3). Tajima’sDvalue of Group 2 was positive and Fu’sFsvalue was negative but not significant (Table 5), indicating that these populations had undergone weak expansion. A smallrvalue, an insignificantSSDvalue and the increased effective population size (Table 5) supported population expansion though the multi-peak results of observed mismatch distributions. According to the value of τ (Table 5), expansion of Group 2 occurred at 0.03 Ma,i.e., the last interglacial period (LIG). Tajima’sDand Fu’sFsvalues of Group 4, Group 6 and Group 7 (Table 5) indicated that these populations had also experienced population expansion, which were consistent with the unimodal results of the observed mismatch distributions, with expansion occurring at 0.0194 Ma, 0.0246 Ma and 0.0823 Ma, respectively.

Fig. 2 Phylogenetic tree (A) and haplotype network (B) based on combined dataset (COI+COII+ND1+ITS)of Sigmella biguttata populationsShort bars in the haplotype network represent mutation steps, the small black dots represent the missing haplotype (Hap), and the haplotype circle size denotes the number of sampled individuals. Values near the node indicate the bootstrap values of the phylogenetic tree.
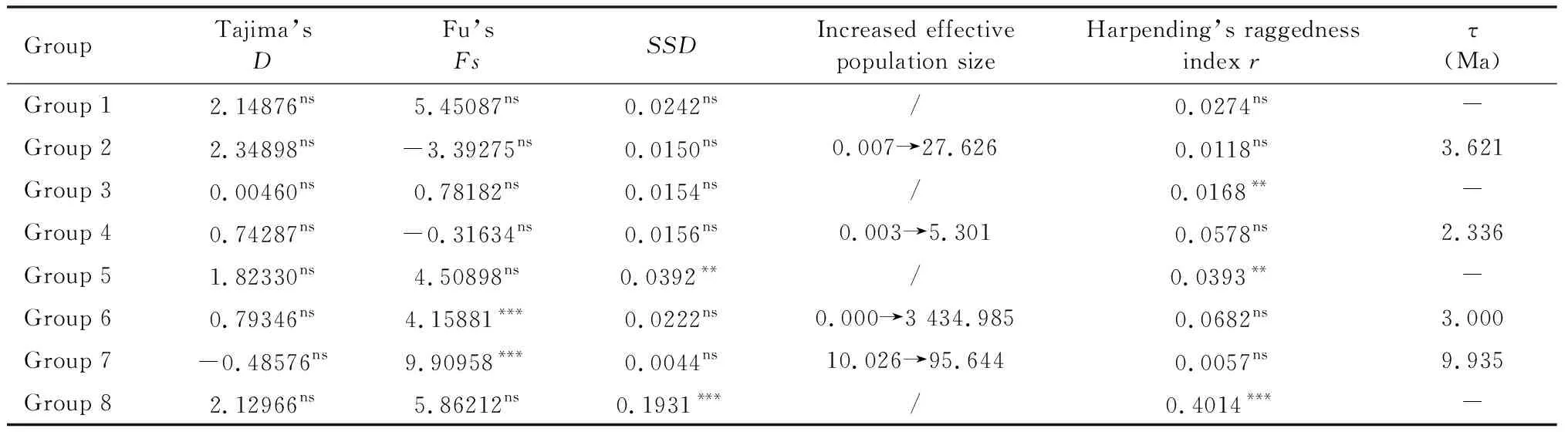
Table 5 Parameter statistics of neutrality and mismatch distribution tests for each group of Sigmella biguttatapopulations based on combined dataset (COI+COII+ND1+ITS)

Fig. 3 Mismatch distribution analysis diagram of each group of Sigmella biguttata populationsbased on combined dataset (COI+COII+ND1+ITS)A-H: Group 1-Group 8, respectively.
3.4 Divergence time estimation
The results of divergence time estimation (Fig. 4) showed that the split between HiN population (lineage I) and mainland China populations (lineages II and III) was dated at 0.5391 Ma [95% confidence interval (CI): 0.1756-1.9949] during the period of Mid-Pleistocene. Lineage II and lineage III diverged at about 0.4512 Ma [95% CI: 0.1369-1.6424] during the period of Mid-Pleistocene. The split within lineage II (between Group 2 and 3) was dated at 0.3249 Ma [95% CI: 0.1039-1.1998] during the period of Mid-Pleistocene (Fig. 4). Most of the divergence time among groups of lineage III occurred at 0.3981-0.1544 Ma (Fig. 4).

Fig.4 Phylogenetic time-tree of Sigmella biguttata populations inferred from the combined dataset (COI+COII+ND1+ITS) using the nucleotide substitution rate of mitochondrial gene COIBlue horizontal bars represent 95% confidence interval estimates for node ages. The value near the node shows the divergence time (Ma).
4 DISCUSSION
4.1 Genetic diversity and differentiation analysis
Our genetic diversity analysis showed that Group 1 (HiN population) had the highest genetic diversity (Hd=0.94466,π=0.00932) (Table 3). Hainan Island has a warm and humid climate, abundant rainfall and suitable altitude (Yan, 2006), which provides a suitable habitat forS.biguttataand helps to improve the genetic diversity. However, Group 4 (JXLS population) exhibited the lowest genetic diversity (Hd=0.75200,π=0.00049) (Table 3), so we speculated that Lushan Mountain is mainly a scenic tourist area and the ecological environment has been greatly damaged, which had led to the habitat destruction then the decrease in genetic diversity as the wild rodent,Calomysmusculinus(Chiapperoetal., 2011). The haplotype network analysis (Fig. 2: B) showed that there was no shared haplotype by a total of 139 haplotypes, indicating that significant genetic differentiation has occurred in different populations due to geographical barriers.
4.2 Phylogenetic and divergence time
The phylogenetic analysis supported a three-lineage including eight groups, showing the clear phylogeographic structure ofS.biguttata. The substitution rate ofCOIgene had been successfully used to infer the time of population differentiation in many phylogeographic studies ofHyalessamaculaticollis(Liuetal., 2018) andMatronabasilaris(Xueetal., 2019). No fossil record for calibration and the estimated age for the most recent common ancestor could be applied, so we performed the divergence time estimation based on the substitution rate ofCOIgene in this study.
Firstly, HiN population (lineage I) diverged from mainland populations (lineages II and III) at 0.5391 Ma (i.e., the Mid-Pleistocene), which was in general consistent with the forming of Qiongzhou Strait (0.7-0.1 Ma). Lineage II and lineage III were estimated to diverge about 0.4512 Ma, which coincided with the complete channelization and formation of Yangtze River finished during the Pleistocene period (2.58-0.018 Ma) (Zhengetal., 2017). Lineage II is mainly composed of populations from south of the Yangtze River; however, in addition to populations from north of the Yangtze River, lineage III also contains four populations (JXLS, AHHS, ZJLHS and JSNJ) from south of the Yangtze River. These four populations (JXLS, AHHS, ZJLHS and JSNJ) were located in the junction of the Oriental realm and the Palaearctic realm, where the animals showed a mixed distribution phenomenon (Liu and Zheng, 1997).
The differentiation among each group of lineage III occurred in the Mid-Pleistocene (0.3981-0.1544 Ma). The Quaternary Pleistocene climatic oscillation (2.58-0.01 Ma) has been proved to be the fundamental driving force for differentiation of many species (Gaoetal., 2012; Zhuetal., 2016; Xueetal., 2019). During this period, the climate cycle transitioned, the amount of global ice increased and the sea level dropped, making the global climate cold and dry (Wuetal., 2002), rapid climate change accelerated the differentiation ofS.biguttata.
4.3 Population historic dynamics
Our study showed that three lineages ofS.biguttatamight have taken different pathways in their evolutionary histories. Group 1, Group 3, Group 5 and Group 8 did not experience obvious expansion due to geographical barriers,i.e., the Qiongzhou Strait, Guizhou Plateau and Wuyi Mountain, which blocked the gene communication among populations. In the Pleistocene, climatic oscillation led to species surviving in refuge and then expansion after the glacial periods as the Chinese Hwamei,L.canorumcanorum(Lietal., 2009). Group 2, Group 4, Group 6 and Group 7 experienced population expansion, which occurred during the LIG, and at that time the global average annual temperature was 2-5℃ higher than that today (Zhaoetal., 2011). In the field collection, we discovered thatS.biguttatatended to live in warm and humid environments, which generally helped to promote population expansion. In the LIG, some species had also expanded, such asParusmonticolus(Wangetal., 2013),Emmenopteryshenryi(Zhangetal., 2016) andAdelphocorissuturalis(Zhangetal., 2015).
The above mentioned results indicate that the genetic differentiation and geographic distribution pattern ofS.biguttataare affected by factors such as geographic barriers and climatic oscillation in the Quaternary Pleistocene, then the populations ofS.biguttatafinally form the current geographic distribution pattern.
猜你喜欢
中国三峡(2022年6期)2022-11-30 06:15:20
艺术品鉴(2022年16期)2022-07-09 03:31:08
河池学院学报(2021年1期)2021-07-10 05:14:16
景德镇陶瓷(2021年1期)2021-03-24 05:47:54
世界农药(2019年3期)2019-09-10 07:04:04
热带生物学报(2019年3期)2019-07-29 08:24:38
东方考古(2017年0期)2017-07-11 01:37:50
家教世界·创新阅读(2017年4期)2017-04-26 07:42:20
家教世界(2017年10期)2017-03-25 12:44:55
水动力学研究与进展 B辑(2017年6期)2017-03-14 07:06:38
