
感染GCRV的草鱼miRNA测序与比较分析
2022-03-15 07:45肖调义张秋实熊舒婷
水生生物学报 2022年2期
肖调义 龙 哲 李 彬 张秋实 秦 玲 熊舒婷
(湖南农业大学动物科学技术学院, 长沙 410128)
草鱼(Ctenopharyngodon idellus)是我国传统的淡水养殖鱼类之一, 2019年总产量达553.3×107kg,产量居全国第一[1]。但人工养殖条件下的草鱼易感染草鱼呼肠孤病毒(Grass carp reovirus, GCRV)引起草鱼出血病, 严重制约了草鱼产业的健康发展。GCRV疫苗是防治草鱼出血病的重要手段之一, 但GCRV的多亚型与变异特性限制了疫苗的预防成效[2,3]。近年来逐步兴起的抗病育种探索有了新的研究进展[4], 利用分子标记辅助选育, 可大大提高选择效率, 缩短育种年限, 已有一些水生动物的抗病相关分子被标记, 并利用该标记培育成优良品系[5,6]。生物机体的免疫水平与遗传因素密切相关[7], 汪亚平团队发现中国长江、珠江及黑龙江等不同水系草鱼的GCRV抗病能力存在差异[8], 因此基于草鱼机体自身的GCRV抗性水平差异的特点开展高抗性草鱼的选育是实现草鱼抗病育种的又一重要策略。不仅是表达基因, miRNA也可列入草鱼的主效抗性分子资源的筛选范围[9,10], 可望为高抗性草鱼的分子辅助育种提供理论参考。
miRNA是一类长约22nt的内源性的、进化上高度保守的非编码小RNA单链分子, 通过与靶基因的3′端非翻译区(Untranslated regions, UTR)结合来抑制靶基因的翻译或降解靶基因, 调控生物体的各项生命活动[11]。miRNA调控生命体的大部分基因[12],参与了生物体重要的生命活动, 如个体发育、细胞增殖、分化、凋亡和免疫等[13—15]。病原体的入侵也会影响机体细胞内miRNAs的表达水平, 借助高通量测序分析鉴定功能miRNA是目前比较普遍的方法。近年来, 一系列组学研究表明miRNAs是鱼类抗病相关研究的重要候选功能分子, 参与硬骨鱼类的抗病毒免疫反应[16—18]。Zhang等[19]综述了在一系列感染虹彩病毒rock bream iridovirus (RBIV)-C1的牙鲆(Paralichthysolivaceus)脾脏中筛选出了121个差异表达的miRNAs, 其预测靶基因主要富集在免疫应答、信号转导和细胞凋亡等信号通路中,揭示miRNA在病毒感染过程中参与了天然免疫过程。
草鱼出血病是目前草鱼产业的极大威胁, 草鱼抗病力水平的高低与遗传因素是紧密相关。因此,本研究借助高通量测序分析草鱼感染前后的头肾中的差异表达的miRNAs, 筛选出了一些候选功能miRNA分子, 并了解其在各免疫器官中的表达模式, 旨在为草鱼的抗病毒分子机制的研究提供理论补充, 为高抗性草鱼的选育及分子设计育种工作提供可参考的信息。
1 材料与方法
1.1 实验动物
本实验的实验草鱼均来自浏阳乌龙渔场, 选取规格7—8 cm左右的6月龄、无病无伤、未感染过GCRV的当年鱼种进行攻毒, 于室内养殖系统中进行攻毒, 养殖水温为恒定28℃。
1.2 病毒毒株及GCRV侵染
草鱼体内攻毒实验所用病毒为草鱼呼肠孤病毒Ⅱ型(GCRV-Ⅱ), 长江所曾令兵实验室馈赠。取30尾草鱼至养殖系统暂养2周(分2组, 对照组10尾,实验组20尾)。攻毒时, 实验组中每100 g草鱼注射毒株1 mL, 对照组中的每尾草鱼注射同等剂量的PBS缓冲液。
1.3 样本采集及小RNA文库构建和测序
待攻毒后的草鱼出现出血症状、濒临死亡时,分别采集对照组及处理组出血症状的草鱼头肾, 每组各采8份组织样品, 迅速置于液氮速冻, 而后放置于-80℃。分别提取对照组及攻毒组的头肾组织总RNA, 将每组中的8份RNA样品等量混合, 分别构建4个重复的miRNA测序文库, 小RNA测序文库制备采用TruSeq Small RNA Sample Prep Kits (Illumina公司, 美国), 而后使用 Illumina Hiseq2000/2500对构建好的文库进行测序, 测序读长为单端 1X50 bp。随后小RNA文库的Solexa测序的工作由杭州联川生物技术股份有限公司完成。
1.4 生物信息学分析
测序得到的原始数据为图像文件经过base calling, 得到以fastq格式存储的结果文件, 称为raw reads, 包含reads的序列及测序质量信息, 为了保证信息分析的质量, 必须对 raw reads 进行质控处理,使用FastQC软件去除原始数据中有带接头的、低质量的垃圾 reads, 得到clean reads, 其中5′接头序列为: 5′-GTTCAGAGTTCTACAGTCCGACGATC-3′,3′接头序列为: 5′-TGGAATTCTCGGGTGCCAA GG-3′; 利用ACGT101-miR软件 (LC Sciences, Houston, Texas, USA)进行后续的一系列分析, 先对clean reads进行长度过滤, 筛选保留碱基长度在18—26 nt的序列(一般来说动物的miRNA长度区间为18—26 nt), 将筛选得到的数据与miRBase22.0数据库进行比对, 再与草鱼的基因组(http://bioinfo.ihb.ac.cn/gcgd/php/index.php?page=download)进行比对, 用来鉴定已知的miRNAs, 保守型的miRNA命名参照miRBase数据库; 将没有比对上已知miRNA的剩余序列与mRNA数据库、Rfam数据库(包含rRNA、tRNA、snRNA、snoRNA、miRNA等非编码RNA)和Repbase数据库进行比对, 尽可能地发现并去除其中可能的rRNA、snoRNA、snRNA、tRNA及可能的repeat associate sRNA, 最终获得的序列数据即有效的数据; 将这些有效的数据再跟草鱼基因组进行比对, 过滤掉非基因组的序列, 而后将剩下的miRNA序列用RNAfold软件进行miRNA二级结构的绘制和预测, 留下符合二级发卡结构的miRNA序列, 定义为novel miRNA, 并这些novel miRNA用PC(Predicted Candidate)标记, 注明5p 或 3p臂端位置; 接下来用ACGT101-miR软件将所有鉴定到的miRNAs的表达量进行归一化处理(Norm值)[20], 同时使用t检验(t-test)的算法对这些miRNA进行表达量的差异分析, 检验筛选显著性的参数为P_value,我们将P的阈值设定为0.01, 即当Pvalue<=0.01时,则表示该miRNA在两组样品间的表达存在显著差异。根据差异miRNAs检测结果, 使用R软件中的pheatmap函数进行层次聚类分析, 热图用颜色变化来反应miRNA表达量的变化, 可根据颜色的变化,直观地看出不同样本中miRNA表达趋势的变化。与此同时, 为了检验对照组和处理组中4个样品之间的重复性, 采用Pearson相关性分析将归一化分析的数据进行两组样品之间的重复性分析。将筛选出来的显著差异的miRNAs(DE miRNAs)进行靶基因预测, 使用的预测软件为TargetScan和miRanda,并取2个软件预测的交集。将预测得到的DE miRNAs的靶基因分别进行GO (Gene Ontology)和KEGG (Kyoto Encyclopedia of Genes and Genomes)的富集分析。
1.5 miRNA靶基因功能分析
为挖掘miRNA的下游靶基因, 我们利用TargetScan和miRanda对差异显著的miRNA分子进行了靶基因预测。选取2款软件预测出来的靶基因的交集结果, 作为差异miRNA的最终的预测靶基因,而后根据miRNA与靶基因之间的对应关系, 首先找出了所有差异表达miRNA的靶基因mRNA与注释库中GO(Gene Ontology)对应的数量, 计算得到的Pvalue≤0.05位阈值, 满足此条件的功能定义为miRNA-mRNA关系对显著富集的功能, 同理, 对靶基因进行KEGG(Kyoto Encyclopedia of Genes and Genimics)的功能富集分析。
1.6 RNA提取、逆转录及荧光定量
RNA提取使用Trizol(Invitrogen公司, 美国)法,部分头肾组织RNA用于高通量测序, 另一部分用于后续的实验验证, 逆转录使用miRNA 1ststrand cDNA synthesis kit(by stem loop; Vazyme公司, 中国), miRNA逆转录采用Stem-loop RT引物方法, 同时用snRNA U6作为内参; 荧光定量使用SYBR Green(Vazyme公司, 中国)作为染料进行荧光定量PCR(quantitative real-time PCR)。利用qPCR对miRNA的表达进行相对定量分析, 并利用2-ΔΔCt方法进行miRNA的数据处理, qPCR的体系、程序方法及运算参照之前的操作[13]。
1.7 数据的统计学分析
为了减小后续的候选功能分子的验证工作的难度, 进行定量验证时, 我们缩小了差异基因的筛选范围, 将t-test的检验筛选显著性的参数设置为P_value≤0.01, Foldchange<-1和Foldchange>1.5 log2(Foldchange)<-1且log2(Foldchange)>0.5|log2(Foldchange)|≥1且归一化均值表达量(Norm值)≥500。所有的荧光定量的实验数据处理用graphpad7.1处理, 作图和显著性分析也均在此软件中完成。定量相关实验结果的显示均采用均数±标准差(mean±SD),显著性分析采用的方法为t检验,*P<0.05为差异显著。
2 结果
2.1 miRNA测序数据
为挖掘GCRV应答过程中的候选功能miRNAs,本实验分析了GCRV处理前后草鱼头肾中的miRNAs表达差异, 实验分两组, 每组4个重复。利用 Hiseq 2000 技术平台进行了Solexa测序, 分别获得了11165309和9946325条原始序列读数(Raw Reads)。去除低质量序列、接头序列和短序列后,分别从两个文库中筛选出883407和733032条高质量序列进行下一步的分析。我们在对过滤后的Valid数据的总数(Total)及种数(Unique)进行了长度分布统计, 结果显示, 本次平均每个样品获得了长度约为18—26 nt的短链分子6918762条, 将其长度分布进行统计分析后发现大部分的序列分子都分布在20—24 nt(占比91.76%), 符合Dicer酶切割的典型特征, 其中, 22 nt的小RNA最为丰富, 占比达38.97%。在所有的短链RNA序列中, 22个核苷酸的小RNA最为丰富, 占比达38.97%(图1A), 这也说明了样品测序结果的可靠性。同时为了检测每个组内的4个样品的重复性, 我们检测了8个样品之间的组间关系, 组间关系结果显示, 对照组和处理组的关系界线明确, 同组间的样品相互之间的关系均大于99.4%, 说明了组间的重复好, 可信度高(图1B)。
2.2 差异miRNA的筛选、鉴定及聚类分析
将miRNA与参考基因组比对, 从对照组和处理组中分别鉴定出703和739个成熟的miRNAs, 其中118个miRNAs仅在处理组中可以检测到, 有82个miRNAs只在对照组中检测到(图1C)。通过数据库比对, 我们检测发现671个miRNA为novel miRNA。差异性显著分析结果显示, 当P<=0.01时, 获得46个极显著上调miRNA和88个极显著下调miRNA(图1D)。这表明在攻毒过程中, 一些miRNA的表达启动, 一些miRNA的表达被抑制, 预示miRNA在病毒侵染过程中发挥着各自不同的生物学功能。通过归一化分析差异miRNA的表达量数据。根据样品miRNA表达谱的相近程度, 我们将差异表达极显著的miRNA进行了聚类分析, 并采用lg (Norm值)进行miRNA表达展示来直观反映聚类表达模式。聚类分析显示, 2个测序组中的4个平行样品的重复性非常好(图2), 证实我们的采样规范, 测序结果可信度高。
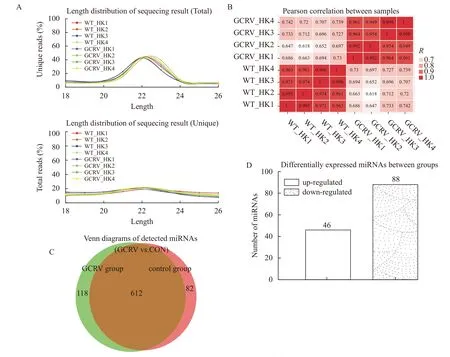
图1 攻毒前后的草鱼miRNA的测序结果分析Fig. 1 Comparing analysis of miRNA sequencing after GCRV challenge in grass carp
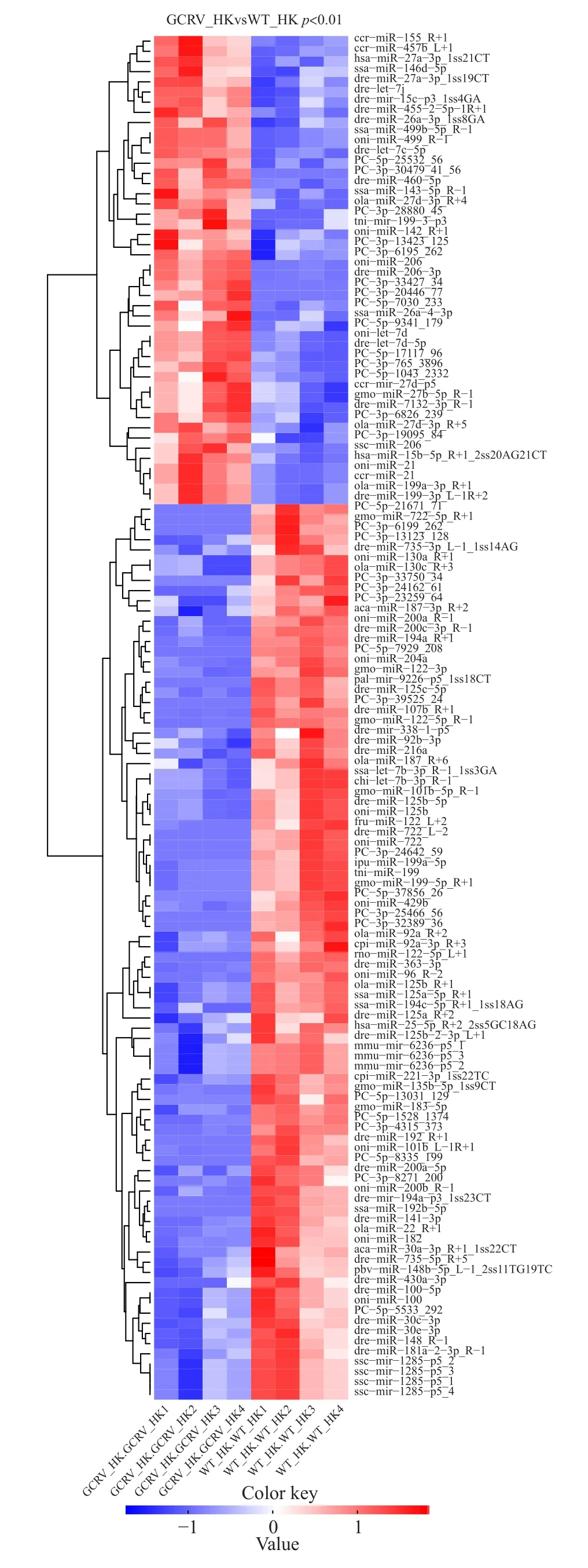
图2 攻毒前后的草鱼DE miRNA热图Fig. 2 Heat map of DE miRNA of after GCRV challenge in grass carp
本实验旨在筛选草鱼抗GCRV的主效功能分子, 因此需尽可能筛选出差异倍数大、表达量相对高的miRNA。于是我们将攻毒前后的miRNA表达量的倍数变化值(FoldChange)和miRNA的归一化均值表达量(Norm值)纳入差异miRNA的筛选参数, 设置筛选参数为|log2(FoldChange)|>=1、Norm值(CON组)>500, 除去成熟序列和表达完全一致的重复miRNA后, 最终获得3个极显著上调miRNA和29个极显著下调miRNA(表1)。

表1 差异表达miRNA列表Tab. 1 List of differentially expressed miRNAs (DE miRNA)
2.3 差异miRNAs的靶基因GO及Pathway富集性分析
在生物体内, miRNA主要通过靶向抑制mRNA发挥其生物学功能, 我们将筛选得到的32个DE miRNAs进行了靶基因预测, 同时将预测得到的所有靶基因进行GO和KEGG的富集性分析。GO功能注释分3大类, 分别是注释基因的分子功能MF(Molecular function)、所处的细胞位置CC(Cellular component)及其参与到的生物过程BP(Biological process)。通过GO注释的结果了解DE miRNA靶基因的功能分布。图3A列出了排名前20的差异miRNA靶基因的GO富集, 富集的基因分别分布在分子功能MF中的转移酶活性“Transferase activity”、共转运体活性“Symporter activity”、钠通道调节器活性“Sodium channel regulator activity”、DNA结合转录因子活性“DNA-binding transcription factor activity”、激酶活性“Kinase activity”、 蛋白激酶抑制因子活性“Protein kinase inhibitor activity”、C-XC趋化因子受体活性“C-X-C chemokine receptor activity”, 生物过程BP中的体节发生过程“Somitogenesis”、多细胞生物发育过程“Multicellular organism development”、DNA转录调控过程“Regulation of transcription, DNA-templated”、凋亡调节过程“Regulation of apoptotic process”、磷酸化过程“Phosphorylation”、通过JAK-STAT途径负调控受体信号通路“Negative regulation of receptor signaling pathway via JAK-STAT”、细胞因子调控的信号通路“Cytokine-mediated signaling pathway”及细胞位置CC大类中的线粒体呼吸链复合物Ⅱ, 琥珀酸脱氢酶复合物(泛素) “Mitochondrial respiratory chain complex Ⅱ, succinate dehydrogenase complex(ubiquinone)”、细胞膜“Membrane”、 闰盘“Intercalated disc”、细胞膜完整成分“Integral component of membrane”及内质网膜“Endoplasmic reticulum membrane”等。
图3B显示了排名前20的差异miRNAs靶基因富集通路, 包括缬氨酸、亮氨酸和异亮氨酸降解(泛素介导的蛋白水解, Valine, leucine and isoleucine degradation; Ubiquitin mediated proteolysis)、TGF-β信号通路(TGF-beta signaling pathway)、牛磺酸和低牛磺酸代谢(Taurine and hypotaurine metabolism)、RIG-I受体信号通路(RIG-I-like receptor signaling pathway)、丙酮酸盐代谢(Pyruvate metabolism)、丙酮酸代谢(Propanoate metabolism)、PPAR信号通路(PPAR signaling pathway)、p53信号通路(p53 signaling pathway)、糖基磷脂酰肌醇通路(Glycosylphosphatidylinositol)、糖鞘脂生物合成(Glycosphingolipid biosynthesis-lacto and neolact)、糖酵解/糖异生(Glycolysis/Gluconeogenesis)、FoxO信号通路(FoxO signaling pathway)、叶酸生物合成(Folate biosynthesis)、丁酸甲酯代谢(Butanoate metabolism)、不饱和脂肪酸的生物合成(Biosynthesis of unsaturated fatty acids)、β-丙氨酸代谢(beta-Alanine metabolism)、上皮细胞的细菌入侵(Bacterial invasion of epithelial cells)、黄曲霉毒素生物合成(Aflatoxin biosynthesis)及脂肪细胞因子信号通路(Adipocytokine signaling pathway)。

图3 差异miRNA靶基因的GO和KEGG富集Fig. 3 GO and KEGG enrichment of DE miRNA target genes
2.4 差异miRNAs的验证
为验证测序结果的准确性, 我们根据对照组(control, CON)的归一化数据Norm值的高低, 将这些DE miRNA分为低、中、高表达的三组, 分别在自行界定的低(500<Norm<600)、中(600<Norm<1000)和高(Norm >1000)3个表达量梯度中选择了12个miRNA用于qPCR验证[miR-96、miR-194ap3_1ss23CT为低表达, miR-155、miR-722、miR-200家族成员(miR-200a/b/c、miR-141)为中表达量组, miR-122-5p、miR-194a、miR-100-5p和miR-21为高表达]。结果显示这12个miRNA的表达趋势与miRNA测序的结果是非常吻合的, miR-96、miR-194a-p3_1ss23CT、miR-722、miR-200家族成员(miR-200a/b/c、miR-141)、miR-122、miR-100-5p和miR-194a等在GCRV处理后显著下调, miR-21、miR-155在GCRV处理后显著上调(图4), 与测序结果非常吻合, 再次确认miRNA测序数据的高准确性和高可信度。值得注意的是, miR-21在GCRV感染组中的头肾中表达量最高, miR-194a在两个比较组中的差异倍数最大, 达到48倍之高。
2.5 DE miRNAs的组织表达分析
为筛选潜在的免疫相关的功能miRNA, 我们筛选了一些候选DE miRNA继续比较分析其在各组织间的表达分布, 旨在探索候选miRNA在肝脏、脾脏、肾脏、头肾、皮肤和鳃等免疫器官中的表达情况。我们挑选的差异显著miRNA如下: miR-200家族成员在垂体中表达量最高, 其次在免疫组织肾脏、皮肤和鳃等组织中有较高表达; miR-100在肌肉中表达量最高; miR-21在脾脏中表达量最高; miR-722在肝脏中表达量最高; miR-194a在肠道中表达量最高, 其次是肝脏和肾脏等组织中; miR-122在肝脏中表达量最高, 其次是脾脏和皮肤等(图5)。

图5 DE miRNA的组织分布Fig. 5 Expression pattern of DE miRNAs in tissues
3 讨论
miRNA在硬骨鱼类的病毒感染过程中发挥潜在重要功能, 许多miRNA会通过靶向作用免疫分子参与调控水产动物的免疫过程[16], 一些功能miRNA在病毒感染过程中可通过调控鱼类的病毒识别受体及下游信号转导中参与的免疫相关基因来及时地调控鱼类的抗病毒天然免疫反应。Najib等[21]在感染病毒性出血性败血症病毒(Viral hemorrhagic septicemia virus, VHSV)的牙鲆头肾组织中鉴定了63个差异miRNA并预测了310个靶基因, 包括IL1B、IRF3、IRF5、IRF7、IL8R和Mx等天然免疫相关分子; 牙鲆在感染巨细胞病毒后, 被诱导上调的 miR-731可通过抑制 IRF7 的表达而减弱Ⅰ型 IFN 反应, 从而有利于病毒自身的复制[22];在棒状病毒感染鮸巨噬细胞过程中, 宿主细胞miR-3570 的表达显著上调, 可靶向调节MAVS介导的 NF-κB 和 IRF3 信号通路, 抑制Ⅰ型 IFN 和抗病毒因子的产生从而促进病毒快速增殖[23]; miR-144可靶向作用traf6直接破坏IRF7的转录, 抑制IFN反应促进病毒的复制[24]。为挖掘草鱼GCRV侵染中的潜在miRNA-mRNA调控网络, 我们对DE miRNA的预测靶基因进行GO和KEGG的富集性分析。GO功能注释显示在分子功能MF中的DNA结合转录因子活性、激酶活性、蛋白激酶抑制因子活性及C-X-C趋化因子受体活性, 生物过程BP中的DNA转录调控过程、凋亡调节过程、磷酸化过程、通过JAK-STAT途径负调控受体信号通路及细胞因子调控的信号通路等均与免疫调控过程紧密相关。与此同时, KEGG的功能富集结果发现包括缬氨酸、亮氨酸和异亮氨酸降解(泛素介导的蛋白水解)、TGF-β信号通路、RIG-I受体信号通路、PPAR信号通路、p53信号通路(p53 signaling pathway)及FoxO信号通路等免疫相关信号通路均发生明显富集。因此, 下一步的研究中可将这些免疫相关通路中的靶基因进行简单的筛选和功能验证。
通过缩小遴选范围, 我们获得了29个下调及3个上调共32个显著DE miRNA, qPCR结果显示12个挑选的miRNA的表达均与测序结果相吻合(图4),不仅从实验上证实了测序结果的可靠性, 同时也预示了测序结果中筛选出来的这32个miRNA可能参与了GCRV应答的免疫调控, 是潜在的功能候选miRNA, 提示我们在头肾中表达量高和变化倍数差异比较大的DE miRNA均具有继续研究的价值。在牙鲆、石斑鱼(Epinephelusspp)、乌鳢(Channa argus)、虹鳟(Oncorhynchus mykiss)、大西洋鲑(Salmo salar)、大黄鱼(Larimichthys crocea)、鮸(Miichthys miiuy)等不同鱼类的不同病毒侵染表达谱分析中发现miRNA-21、miRNA-155和miRNA-100的表达均发生了显著变化, 为保守的DE miRNAs, 同时它们均参与了高等脊椎动物的免疫应答调控[25]; 在细菌感染过程中, 鮸miR-21通过IRAK4介导的NF-κB信号通路调节炎症反应并且其在TLR信号通路调节中起重要作用[26]; Hua等[27]在草鱼肾脏细胞系和斑马鱼(Danio rerio)成鱼进行miRNA的过表达, 提示了miR-155可通过靶向作用mIg的适应性免疫中发挥作用; 虾的miR-100通过靶向胰蛋白酶基因mRNA而发挥抗凋亡作用[15]; 在本次草鱼GCRV感染下的差异分析中, 这3个miRNA都发生了显著差异。综上, miR-21、miR-100及miR-155可作为下一步研究对象的重要候选分子。
为了进一步筛选免疫分子, 我们对部分DE miRNA进行了组织表达分析, 旨在分析各DE miRNA在免疫相关组织中的表达情况。结果显示miR-722和miR-122均在肝脏中表达量最高, 这与大西洋鲑中的结果是一致的[28]; miR-194a在肠道中表达量最高, 且miR-194a在头肾中攻毒前后的表达差异达48倍之高, 牙鲆miR-194a可调控IRF7及IFNI等来调控牙鲆的免疫应答[29], 提示miR-194a也是一个潜在的免疫分子; miR-200家族成员在垂体中表达量最高, 与此同时其在免疫组织肾脏、皮肤和鳃等组织中有较高表达, 提示了miRNA家族成员在体内生物学功能中的保守性; 草鱼出血病的主要表现为鳃、皮肤、肌肉及内脏充血, 而miR-200家族在这些组织中也有高表达, 并且均是GCRV应答中的显著差异分子, 预示着miR-200在草鱼的GCRV应答中发挥潜在重要功能。半滑舌鳎(Cynoglossus semilaevis)中的结果也显示miR-200a和miR-200b可能在免疫调控中发挥功能[30], 揭示了miRNA在种间的功能保守性。值得注意的是, 组织定量结果显示miR-200家族均在垂体中有着超高表达, 这与此前我们在同是鲤科鱼类的斑马鱼中的研究结果类似:miR-200家族成员(miR-200a/b/miR-429a)被证实在斑马鱼雌鱼的生殖发育过程中发挥至关重要的功能[31,32], 这些结果说明了miR-200表达特性在种间具有一定的保守性, 同时还提示我们miR-200不仅可作为免疫方向的研究内容, 同时miR-200在草鱼的生殖发育方面的功能可作为一个新的研究点。
综上, 本研究利用高通量测序对GCRV处理前后的草鱼头肾组织进行了miRNAs的差异分析, 并获得了29个下调及3个上调共32个基因极显著DE miRNAs, 通过进一步的分析, 挖掘出了miR-21、miR-100、miR-155、miR-722、miR-122、miR-194a及miR-200等7个miRNA在免疫调控中发挥潜在重要功能, 为草鱼抗病相关分子的研究及抗病草鱼的选育工作提供研究思路。
猜你喜欢
当代水产(2022年7期)2022-09-20
理财周刊(2022年4期)2022-04-30
当代水产(2022年3期)2022-04-26
昆明医科大学学报(2022年1期)2022-02-28
中国生殖健康(2020年4期)2021-01-18
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
当代水产(2019年9期)2019-10-08
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
