
GC-MS法测定盐酸苯海索原料药中遗传毒性杂质氯代环己烷
2022-03-13 07:26:08殷丽宁胡一桥詹新安
中国药科大学学报 2022年1期
殷丽宁,张 煜,胡一桥,詹新安
(1南京大学生命科学学院,南京 210093;2海正药业(杭州)有限公司,杭州 311404)
药物中潜在遗传毒性杂质的过量残留可能会诱发肿瘤,影响用药安全[1-2]。2020年版《中华人民共和国药典》中新增了“遗传毒性杂质控制指导原则”[3],足见遗传毒性杂质研究在药物质量研究与控制中日益重要。 盐酸苯海索(trihexyphenidyl hydrochloride,THP)是治疗帕金森病的代表性药物[4]。为给THP制剂提供安全质量可控的原料药,对THP 原料药合成工艺进行调研(图1)[5],发现其合成物料之一氯代环己烷(chlorocyclohexane,CCH)具有遗传毒性警示结构,需对其在THP 原料药中的残留量进行检测和控制。根据国际人用药品注册技术协调会(ICH)指导原则[6],对于服药时间长达10 年以上的THP 药物,可接受的毒理学关注阈值为每人每天1. 5 μg,结合用药说明中的最大日剂量20 mg,则CCH 在THP 药物中的可接受杂质限度为75 μg/g(1. 5 μg/20 mg),远低于一般杂质控制限度,因而需要建立一种高灵敏度的分析方法用于THP中CCH的痕量测定。
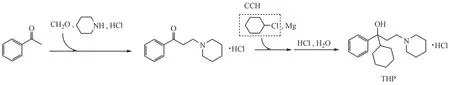
Figure 1 Synthetic route of trihexyphenidyl hydrochloride (THP). Chlorocyclohexane (CCH) with potential genotoxicity is involved via a Grignard reaction in THP preparation
CCH 作为一种挥发性小分子化合物,优先考虑气相色谱检测。为实现对THP中CCH 的精准定量,需尽可能地降低其定量限浓度,如以ICH 指导原则下CCH 限度水平的10% 为基准时,其定量限度为7. 5 μg/g。然而Liu 等[7]发现氯代烷基化合物对常规火焰离子化检测器(FID)及电子捕获检测器(ECD)响应很弱,其检出限至少应达到40 μg/g,对应其定量限应高于120 μg/g(相对于10 mg/mL供试品溶液),远不能满足CCH 的定量限要求。尽管提高THP 样品溶液浓度可以满足供试品的测定需求,但是高浓度样品会污染进样衬管进而影响检测结果的重复性和准确性,甚至缩短气相色谱柱的使用寿命[8]。目前,尚未有文献报道在7. 5 μg/g 的浓度水平下定量检测THP 中CCH 含量的方法,且各国药典中均未收录THP 药物中遗传毒性杂质的定量分析方法。
鉴于此,本研究建立了一种高灵敏度的GCMS 法用于THP 原料药中CCH 的痕量检测。通过筛选合适的内标化合物、样品配制方式和色谱参数,最终实现最佳灵敏度和最优检测效率。该方法经过全面的方法学验证评价,可应用于THP 原料药中CCH 的痕量检测,为THP 药物的质量控制提供了参考。
1 材 料
1. 1 药品与试剂
THP原料药(批号:1804001/1804002/1804003,常州康普药业有限公司);CCH 对照品(纯度:98. 0%,批号:YX6TC-IK,日本东京TCI化成工业发展有限公司);正壬烷(≥99. 5%,批号:F1820086,上海阿拉丁生物化学技术有限公司);乙腈(色谱级,美国霍尼韦尔公司);甲醇(质谱级,德国默克制药与生物技术公司);乙醇(色谱级,南京化学试剂股份有限公司);其他试剂均为市售分析纯。
1. 2 仪 器
GC-MS-QP2010 SE 气相色谱仪、AUW220D 电子天平(日本岛津公司)。
2 方 法
2. 1 色谱和质谱条件
2. 1. 1 色谱条件 色谱柱:SH-RXI-5SIL MS 毛细管柱(0. 25 mm × 30 m,0. 25 μm);进样口温度:180 ℃;柱温:以60 ℃恒温维持6 min;载气:高纯氦气;流速:2 mL/min;进样量:1. 0 μL;进样分流比:10∶1。
2. 1. 2 质谱条件 电子轰击源:EI 离子源;检测器电压(相对于调谐结果):0. 3 kV;离子源温度:200 ℃;气相色谱-质谱接口温度:240 ℃;溶剂延迟时间为1. 3 min;检测模式:SIM;CCH 的检测离子:m/z82;内标正壬烷的检测离子:m/z85。
2. 2 溶液配制
2. 2. 1 空白溶剂 取乙醇为空白溶剂。
2. 2. 2 内标溶液 取内标化合物正壬烷适量,用乙醇溶解并稀释,制成质量浓度约为8 μg/mL 的内标储备溶液;精密量取内标储备溶液1. 0 mL,置10 mL 量瓶中,乙醇稀释制成质量浓度约为0. 8 μg/mL的内标溶液。
2. 2. 3 CCH 对照品溶液 取CCH 对照品适量,用乙醇溶解并稀释,制成质量浓度约为6 μg/mL 的CCH 储备液;精密量取CCH 对照品储备溶液1. 0 mL,置10 mL 量瓶中,乙醇稀释制成质量浓度约为0. 6 μg/mL的CCH对照品溶液。
2. 2. 4 混合对照品溶液 分别移取内标储备溶液和CCH 对照品储备溶液各1. 0 mL到10 mL量瓶中,乙醇稀释制成每毫升中约含0. 8 μg 内标化合物和0. 6 μg CCH的混合对照品溶液。
2. 2. 5 供试品溶液 精密称定THP原料80 mg于10 mL 量瓶中,精密移取1. 0 mL 的内标储备溶液于同一量瓶中,加入乙醇溶解并定容。
2. 2. 6 加杂供试品溶液 精密称定THP原料80 mg于10 mL 量瓶中,分别移取内标储备溶液和CCH对照品储备溶液1. 0 mL 于同一量瓶中,加入乙醇溶解并定容。
3 结 果
3. 1 系统适用性
按“2. 2”项配制混合对照品溶液,照“2. 1”项下色谱条件注入GC-MS 色谱仪进行检测,重复测定6 次并计算CCH 峰面积与内标峰面积比值的相对标准偏差(RSD)。结果显示,待测物CCH 和内标正壬烷峰面积比值的RSD 为1. 56%,小于可接受标准5. 0%,表明分析方法系统适用性结果良好。
3. 2 专属性
按“2. 2”项下方法配制空白溶剂和混合对照品溶液,照“2. 1”项下条件注入GC-MS 色谱仪进行检测,记录色谱图(图2-A),空白溶剂基线平稳,在CCH 和内标正壬烷的保留时间处未检测到信号,对样品测定无干扰。
3. 3 检测限和定量限
分别以3∶1 和10∶1 的S/N 获得LOD 和LOQ。基于此,计算得出LOD为17. 92 ng/mL(2. 24 μg/g),而LOQ 为59. 72 ng/mL(7. 47 μg/g)。LOQ 远小于可接受摄入量极限(75 μg/g)。图2-B 和2-C 分别显示了LOD和LOQ浓度的分析物的质谱图。
3. 4 线性关系
在浓度水平LOQ(10%)至250% 限度范围内选取6 个浓度,照“2. 1”项下色谱条件注入GC-MS色谱仪进行线性验证,将测得的响应信号(CCH 与内标峰面积的比值)与CCH 浓度用最小二乘法进行线性回归统计计算,得到回归方程和相关系数。结果表明,CCH 的线性方程为:y= 1. 588 5x+0. 011,在59. 72 ~ 1 493 ng/mL(7. 47~186. 6 μg/g)浓度范围内呈现良好的线性相关性(r= 0. 999 9)。
3. 5 准确性
向供试品本底溶液中加入相当于限度浓度指标的10%、50%、100%、150% 4个浓度水平的杂质,各浓度配制3 份,照“2. 1”项下色谱条件注入GCMS 色谱仪进行检测并计算杂质回收率。结果显示,从低到高的4 个浓度水平下,CCH 的平均回收率依次为99. 82%、99. 90%、100. 1% 和100. 4%,总体回收率的RSD 为1. 29%,方法准确度良好。
3. 6 精密度
不同分析人员于不同日期按“2. 2”项下方法平行配制6 份加杂供试品溶液,照“2. 1”项下条件注入GC-MS 色谱仪进行检测。结果显示,6 份加杂供试品溶液中CCH 含量的RSD 为0. 96%,12 份加杂供试品溶液中CCH 含量的RSD 为1. 69%,说明该方法的日内、日间精密度良好(RSD ≤5. 0%)。
3. 7 耐用性
通过改变柱温(± 2 ℃)、柱流量(± 0. 05 mL/min)、进样口温度(± 5 ℃)、接口温度(± 5 ℃)来评估。按“2. 2”项配制CCH 对照品溶液和加杂供试品溶液,在上述变动条件下进样分析,记录色谱图并计算CCH 的回收率。结果(表1)显示方法具有较好的耐用性。

Figure 2 Results of specificity and limit. Representative mass chromatograms of (A) diluent and standard solution, (B) limit of detection (LOD), and(C) limit of quantitation (LOQ)
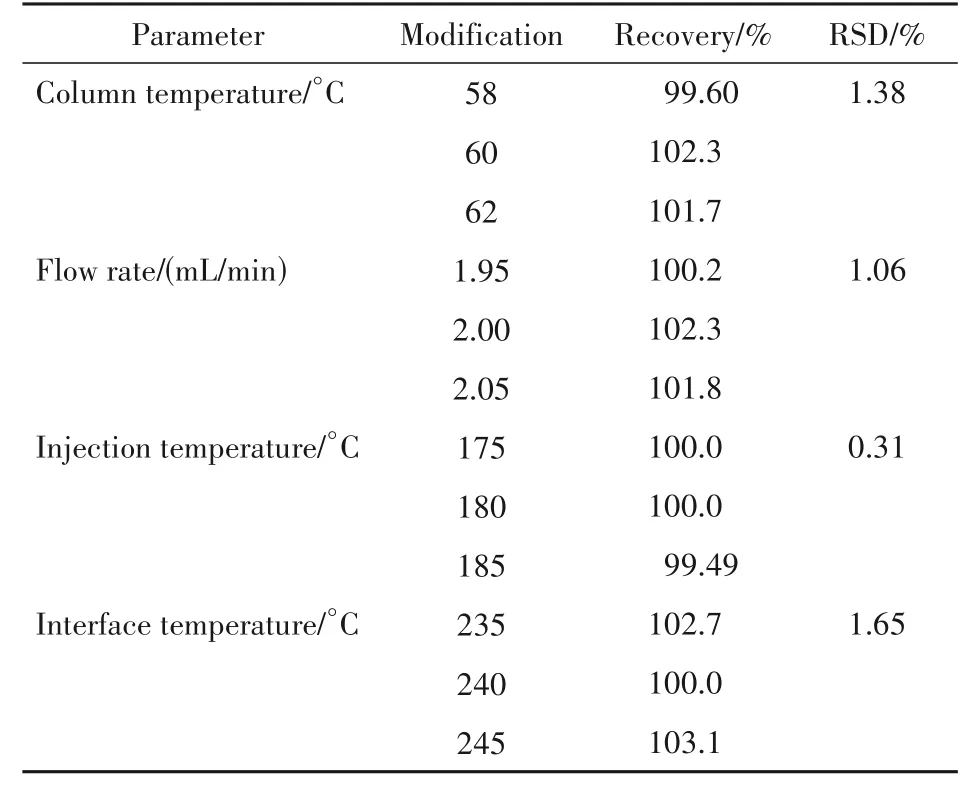
Table 1 Robustness data expressed as the recovery of CCH
3. 8 样品测定
对3 批THP 原料药(批号:1804001、1804002、1804003)中的潜在基因毒杂质CCH 含量进行检测,按“2. 2”项下方法平行配制6份供试品溶液,照“2. 1”项下条件采用GC-MS 色谱仪进行检测。结果显示,3 批THP 原料药均未检出CCH 残留,该药品中遗传毒性杂质含量远低于可接受限度标准要求。
4 讨 论
4. 1 CCH的定性研究
利用GC-MS 对m/z50~m/z150 范围内的正离子进行全面扫描从而对CCH 进行定性检测。根据标准质谱谱库NIST17比对结果及质谱裂解图最终确定了CCH 的结构。图3 显示了CCH 的3 个主要碎片离子,包括m/z67,m/z82和m/z55并推测其片段结构。在这些离子中,选用m/z82作为选择性离子监测(SIM)模式中的特征定性离子,用于CCH 定量分析。

Figure 3 EI mass spectrum and speculative fragment structures of CCH
4. 2 内标化合物的筛选
内标法是用于遗传毒性杂质痕量分析的理想定量方法,可校准和消除进样量差异、色谱条件变化误差、检测器条件波动等对于分析结果的影响,从而提高定量检测方法的稳定性和准确度[9]。内标化合物的筛选是内标法应用的关键。气相色谱中物质的保留时间与沸点相关,因此首选考察与待测物CCH 沸点(142 ℃)相近的化合物作为内标的适用性。初步选定的内标化合物包括正辛烷(沸点126 ℃)、乙酸丁酯(沸点126 ℃)和正壬烷(沸点151 ℃)。从图4 中可以看出,正壬烷与CCH的保留时间接近且可较好分离,而正辛烷和乙酸丁酯的峰与CCH 的峰相距甚远。此外,正壬烷峰形尖锐且特征定性离子为m/z85,不影响CCH 定量。因此,正壬烷被选为内标化合物。
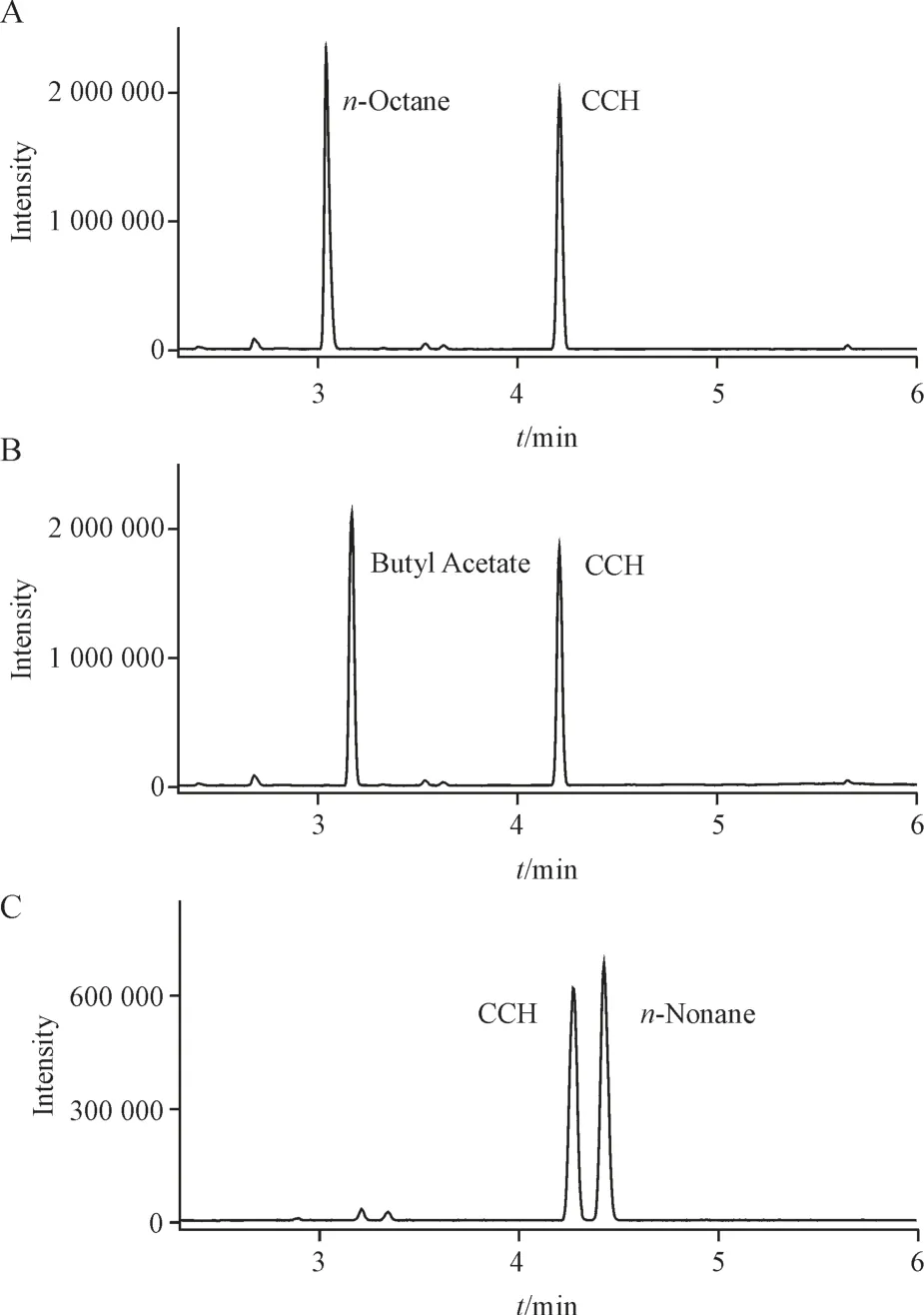
Figure 4 Mass chromatograms of 3 potential ISTDs (internal stan⁃dards). (C) n-nonane had better peak shape and more similar retention time with CCH compared with (A) n-octane and (B) butyl acetate
4. 3 溶剂筛选
溶剂选择是样品配制方法的关键,合适的溶剂不仅对样品有良好的溶解性,而且对样品分析无干扰。本研究从溶解性能和检测干扰两个角度考察了常用挥发性溶剂乙腈、甲醇和乙醇的对于本方法的适用性。
首先考察空白溶剂是否会对检测造成干扰。将乙腈、甲醇和乙醇3 种溶剂依次进行GC-MS 分析,色谱结果显示,甲醇杂峰较多且在内标正壬烷的保留时间(4. 4 min)处有一个明显的干扰峰,不适合作为样品配制的溶剂。乙醇和乙腈的基线都较为平稳,未对检测造成干扰,可进行进一步筛选。
其次考察溶解性。分别向乙醇和乙腈溶剂中加入样品检测所需浓度的CCH、正壬烷和THP 原料药,观察溶解情况。结果显示,CCH 和正壬烷在两种溶剂中均能完全溶解;THP 原料药在乙腈中溶解度低于2 mg/mL,无法完全溶解,而在乙醇中表现出良好的溶解度。因此,最终选用乙醇作为样品溶液配制的溶剂。
4. 4 优化GC-MS条件
由于CCH 和内标化合物正壬烷沸点相近(沸程<10 ℃),为实现待测组分的有效分离和良好峰形且简化分析检测流程,最终采用恒温程序而不是升温程序。
基于此,进一步优化GC-MS 条件。为提高方法灵敏度至限度要求水平,分别对关键的分析方法质谱和色谱参数进行优化。以CCH 色谱峰的信噪比(S/N)作为评判灵敏度的量化标准,考察参数包括检测器电压(相对于调谐结果,0. 1 kV、0. 2 kV、0. 3 kV)、气相色谱-质谱接口温度(225 ℃、240 ℃、260 ℃)和恒温程序中的柱温(55 ℃、60 ℃、65 ℃)。
如图5 所示,检测器电压对CCH 响应有显著影响,设置值为0. 3 kV 时功能最佳;240 ℃的界面温度比225 ℃和260 ℃略有优势,信噪比依次提高了15%和27%;当色谱柱柱温保持在60 ℃时,CCH 峰形对称且尖锐,信噪比高且保留时间适中。因此,在GC-MS 条件的系统优化中最终选择了0. 3 kV,240 ℃和60 ℃,3 个色谱参数被证明对于THP 原料药中CCH 的痕量测定具有足够灵敏度。
5 讨 论
本研究建立了一种简单、新颖且效率高的GCMS 分析方法,该方法可用于THP 原料药中遗传毒性杂质CCH 的痕量检测。实验结果证明,该方法具有良好的专属性、线性、准确度、精密度以及耐用性,适用于THP 原料药中遗传毒性杂质的质量控制,同时也为开发其他烷基卤化物的痕量测定方法提供一定的参考。
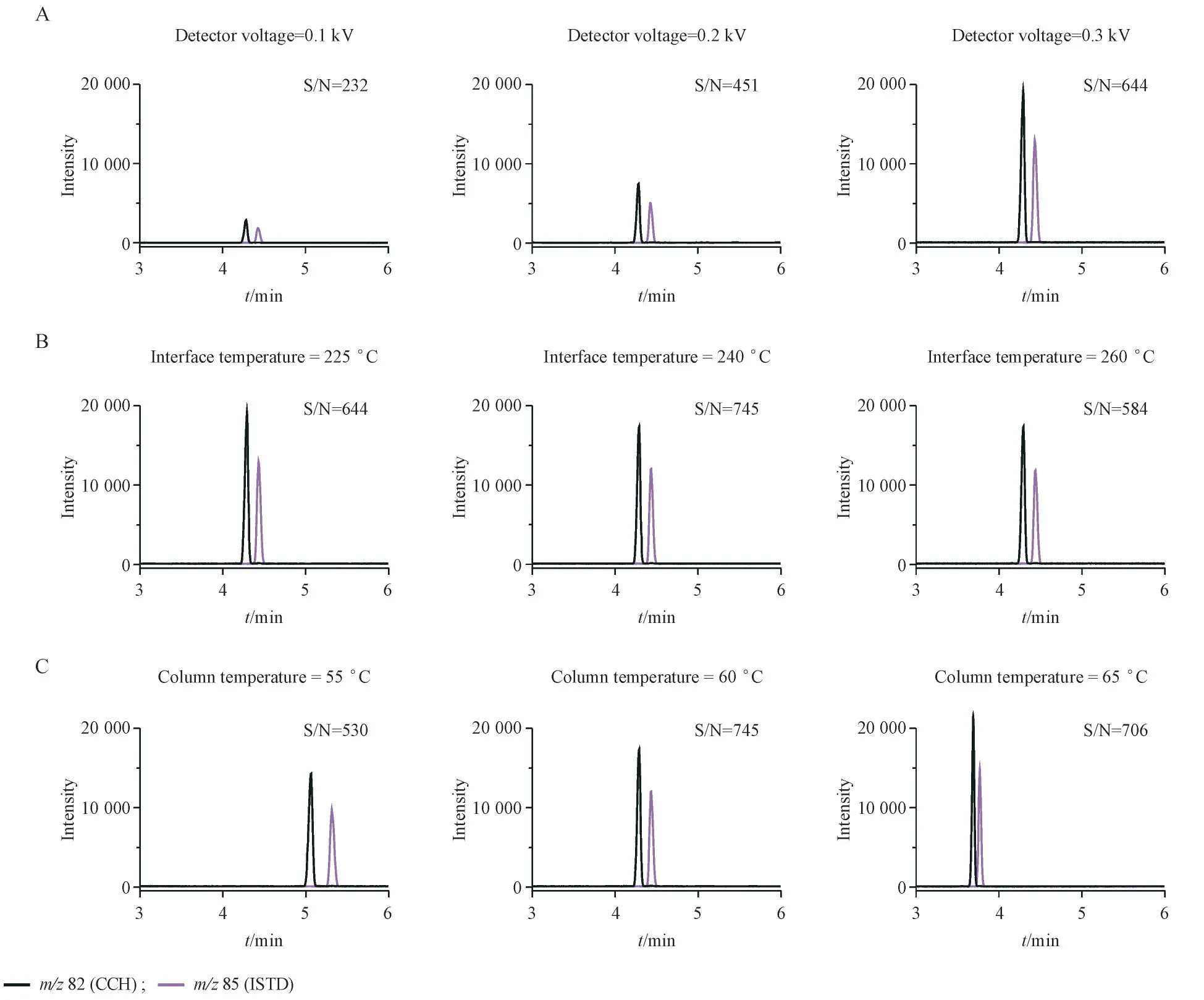
Figure 5 Optimization of GC-MS conditions of detector voltage (A), interface temperature (B) and column temperature (C)
猜你喜欢
云南化工(2021年7期)2021-12-21 07:27:48
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
艺术品鉴(2020年6期)2020-12-06 10:49:08
中国盐业(2018年20期)2019-01-14 01:18:42
中成药(2018年6期)2018-07-11 03:01:32
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
中国粮油学报(2016年5期)2016-01-23 02:45:06
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
