
小烛树蜡和日本木蜡对高油酸葵花籽油凝胶油结晶行为的差异比较
2022-03-10 02:35史逸飞刘春环郑召君刘元法
中国油脂 2022年2期
史逸飞,刘春环,郑召君,刘元法
(江南大学 食品学院,江苏 无锡 214122)
塑性脂肪常被应用于人造奶油、起酥油、巧克力等烘焙或休闲食品中,能够赋予食品良好的结构、口感或风味等。但传统的塑性脂肪往往存在着一定的健康风险,其中大量的饱和脂肪酸和一定的反式脂肪酸可能会诱发如肥胖、心血管疾病、2型糖尿病等疾病[1]。凝胶油被认为可以作为传统塑性脂肪的替代品,主要是通过凝胶因子形成的网络结构固定连续的油相,形成具有稳定性结构的半固态油脂[2]。
目前已有多种方法制备凝胶油,包括结晶法[3]、分子自组装法[4]、乳液模板法[5]及多孔材料吸附法[6]等,其中以结晶法研究最为广泛。结晶型凝胶因子有天然蜡、单甘酯[7]、硬脂酸钠[8]、L-抗坏血酸棕榈酸酯[9]等,其中天然蜡因来源天然、结晶性强、成本低等优点受到广泛的关注[10],常被用作凝胶剂或结晶改良剂[11]。天然蜡主要成分为长链蜡酯、烷烃、游离脂肪酸、脂肪醇、甾醇酯等[12]。目前已有研究将蜂蜡、巴西棕榈蜡、小烛树蜡、葵花蜡、浆果蜡、甘蔗蜡等天然蜡用于凝胶油的制备[13-17],不同天然蜡由于成分和来源的差异具有不同的结晶形态和结构,因而具有不同的凝胶能力和机械性能。了解天然蜡在植物油中的结晶过程是开发新型塑性脂肪,拓展其在食品中应用的必要条件。木蜡也被称为漆蜡,提取自我国传统的经济林漆树的果实,主要成分为脂肪酸酯、少量游离脂肪酸和脂肪醇等,其脂肪酸以棕榈酸、硬脂酸和油酸为主,同时还含有少量的二元脂肪酸,熔点在32~40℃。漆树自中国传入日本后出现品种差异,日本漆树的漆籽含蜡率高于含油率,且日本木蜡比中国木蜡含有更多的花生酸和山嵛酸[18]。作为低熔点的天然蜡,由于成分差异,日本木蜡结晶特性与高熔点蜡不同,因此有必要研究其在植物油中的结晶行为来拓展其应用范围。目前仅有Hwang等[13]对日本木蜡凝胶机理进行研究,但对于其结晶机理并未进行深入探讨。
小烛树蜡提取自美国西南部和墨西哥北部的大戟属灌木叶[19],其主要成分为75%左右的正构烷烃(C31为主)、15%左右的蜡酯以及少量的脂肪酸和脂肪醇[16],是常见的几种天然蜡中唯一以烷烃为主要成分的蜡,熔点在65℃左右,具有独特的结晶特性。
本文对比研究了两种不同熔点的天然蜡(日本木蜡、小烛树蜡)在植物油中的结晶行为,通过流变仪、差示扫描量热仪、偏振光显微镜、X-射线衍射仪和傅里叶红外光谱仪等检测手段对两种蜡基凝胶油的流变学性质、热力学性质、微观结晶形态、纳米层状结构以及分子间作用力等进行表征,系统评价两种蜡的结晶网络结构,以期扩大木蜡在食品中的应用范围,为新型塑性脂肪的开发和应用提供理论依据。
1 材料与方法
1.1 实验材料
小烛树蜡(CLW)、日本木蜡(RLW),天津万科斯有限公司;高油酸葵花籽油,日清奥利友集团。
D2-Advance X-射线衍射仪,德国Bruke公司;DSC3差示扫描量热仪,瑞士Mettler公司;偏振光显微镜,德国Leica公司;ME-12循环水浴锅,德国Julabo公司;DHR-3旋转流变仪,沃特斯中国有限公司;多点磁力搅拌器,美国TA公司;IS-10傅里叶红外光谱仪,美国Nicolet公司。
1.2 实验方法
1.2.1 凝胶油的制备
分别将5 g小烛树蜡和日本木蜡分散在100 g高油酸葵花籽油中,混合物在85℃的多点磁力搅拌器上以400 r/min加热搅拌20 min,直至天然蜡完全溶解,在4℃下冷却保存至少48 h,备用。
1.2.2 流变学分析
将凝胶油样品保持在20℃下分析。使用装有直径为40 mm、间隙1 000 μm平行板的DHR-3旋转流变仪对凝胶油进行流变学测量,采用Peltier系统进行温度测试。在频率为1 Hz、应变振幅范围为0.001%~100%的区域进行振幅扫描,以确定线性黏弹区(LVR),据此在应变为0.01%下进行频率扫描,扫描范围为0.1~10 Hz,记录该扫描范围储存模量(G′)和损耗模量(G″)的变化。采用1 Pa的振荡应力和1 Hz的频率以5℃/min的速率从0℃升温至80℃测量样品随温度变化下的G′和G″。以1~100 s-1的剪切速率进行流动扫描,测试样品的表观黏度变化,并用Herschel Bulkley模型拟合[20],计算流动行为指数(n)。以0.1 s-1300 s、1 s-1200 s、0.1 s-1300 s的低剪切速率和高剪切速率交替实验测试样品黏度的回复性,以第三阶段的峰值黏度与第一阶段的终黏度的比值即触变回复率表示[21]。
1.2.3 热分析
使用DSC3差示扫描量热仪测定凝胶油样品的热性能,用纯铟校准DSC基线和温度。称取3~8 mg样品放入坩埚,并以空坩埚作为对照。首先将样品在80℃下放置10 min直至样品完全熔化消除晶体记忆,随后以5℃/min速率冷却至0℃,分析样品的结晶行为;之后将样品在0℃下放置10 min达到平衡,然后以5℃/min速率加热到80℃,分析其熔化行为。
1.2.4 微观晶体网络形态
使用偏振光显微镜(PLM)获取样品的晶体微观结构和形态。所有样品均于90℃完全熔化30 min后进行点样,置于4℃或20℃培养箱中保存48 h观察。分形维数(Db)由Image J软件v.1.49(NIH,Bethesda, USA)根据以往研究计算[22]。
1.2.5 晶型与晶体多态性分析
采用X-射线衍射仪(XRD)测定样品的长、短间距的衍射图谱。晶体网络分析采用Ni滤波器和Cu-K抗辐射(k=1.540 56 Å,电流40 mA,电压40 kV,固定散度10、1.0、0.1 mm,抗散射和接收缝)。在环境温度下,样品以0.05(°)/min的速率进行1°~30° 的扫描。数据处理和分析使用MDI Jade 6.0软件。
1.2.6 分子间作用力
采用傅里叶红外光谱仪分析凝胶油样品的分子间作用力。将样品放置在待测区,在400~4 000 cm-1的波长范围内获得光谱。从光谱中减去空气背景,并通过OMNIC(Thermo,v8.0)软件进行分析。
1.2.7 数据统计与分析
使用Origin 2018进行统计分析。采用SPSS 26软件进行单因素方差分析(ANOVA)和沃勒-邓肯检验,P<0.05表示差异显著。
2 结果与讨论
2.1 天然蜡基凝胶油的流变学性质(见图1)

在5%添加量下,小烛树蜡和日本木蜡均能够形成在倒置条件下不流动的软固体。从图1A可以看出,在线性黏弹区范围内,两种凝胶油均呈现类固体性质(G′>G″),且两种凝胶油的G′和G″对频率的依赖性很低,代表“强凝胶”的形成[23],反映了凝胶油中存在较强的结晶网络连接[24]。同时,日本木蜡凝胶油的G′和G″均低于小烛树蜡凝胶油的。说明高熔点的蜡制备的凝胶油具有较高的机械强度。由图1B可以看出,两种凝胶油表现出不同的温度转变点(G′=G″),且转变温度与蜡的熔点成正比。在低温下两种凝胶油均表现出固体特征,而随着温度的升高,两种凝胶油的G′不断下降,低熔点的日本木蜡凝胶油首先发生转变,由弹性为主的固态转变为黏性为主的液态,说明凝胶油的结晶网络坍塌。而小烛树蜡凝胶油具有更高的温度稳定性,同时也表示其需要更高的温度才能破坏其内部的结晶网络结构。
由图1C可以看出,随剪切速率增大,两种凝胶油的表观黏度减小。通过Hershel Bulkley模型拟合,日本木蜡凝胶油和小烛树蜡凝胶油的n值分别为0.403和0.230。说明两种凝胶油均表现出剪切变稀行为和假塑性行为(n<1),这表明在剪切过程中会导致样品结晶网络的解体。高熔点的小烛树蜡凝胶油具有较高的表观黏度,表明小烛树蜡凝胶油具有更强的结构稳定性。由图1D可以看出,剪切速率从0.1 s-1增加到1 s-1再降低至0.1 s-1,日本木蜡凝胶油的触变回复率为56.18%,小烛树蜡凝胶油的触变回复率为48.16%,这与其机械强度相反,根据文献[25]报道,这可能是由于小烛树蜡凝胶油形成的结晶网络中的脆性结构致使剪切力克服凝胶油中结晶的布朗运动,使得破碎的结构无法重新组织为一个较为完整的网络结构,而日本木蜡凝胶油在受到较强的剪切力后,在弱剪切力的作用下体系内的结晶逐渐返回到随机分布的状态。两种蜡基凝胶油的机械强度和黏度变化说明,高、低熔点蜡在构建结晶网络时的性质差异,导致了机械强度与回复性表现出相反的结果。
2.2 凝胶油的结晶与熔化行为表征
为进一步分析凝胶油在结晶网络形成过程中的转变信息,通过DSC分析了两种蜡及其凝胶油的结晶和熔化过程,结果如图2所示。表1为各样品对应的熔融/结晶起始温度、峰值温度及焓值变化。
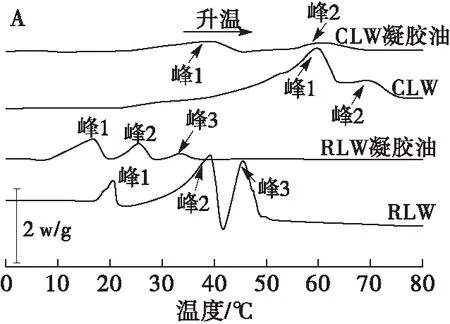

表1 小烛树蜡(CLW)和日本木蜡(RLW)及其凝胶油的熔融/结晶起始温度(Ton)、峰值温度(Tp)和焓值(ΔH)
由图2及表1可见,相同蜡构建的凝胶油与纯蜡的结晶和熔融峰相似,表明蜡在凝胶油的热力学行为中起主导作用。在熔化过程中,小烛树蜡构建的凝胶油与小烛树蜡均存在2个熔融峰,小烛树蜡凝胶油2个峰的焓值分别为2.01 J/g和1.52 J/g,而小烛树蜡纯蜡2个峰的焓值则分别为71.32 J/g和11.09 J/g,液油的加入使Ton向低温区移动,焓值减小。日本木蜡凝胶油及日本木蜡均具有3个熔融峰,其中日本木蜡凝胶油的第1个峰具有最高的焓值(2.72 J/g),后2个峰焓值较低,而日本木蜡则在第2个峰显示出最高的焓值(42.73 J/g),推测是由于日本木蜡的主要成分为脂肪酸酯[18],在形成凝胶油的过程中受到液油的影响[26],从而造成凝胶油焓值减小。与小烛树蜡凝胶油相比,日本木蜡凝胶油熔融峰温更低,在33.59℃即能完全熔化,表明日本木蜡凝胶油形成的网络结构较弱。在结晶过程中,两种蜡与其凝胶油样品均表现出相似的峰型,小烛树蜡凝胶油及小烛树蜡的第1个峰的峰值温度分别为48.07℃和66.98℃,日本木蜡凝胶油及日本木蜡的峰值温度分别为10.99℃和35.27℃,说明纯蜡能够在更高的温度下结晶,液油的加入降低了结晶温度。
2.3 凝胶油的微观结构
2.3.1 微观形态
图3为小烛树蜡凝胶油和日本木蜡凝胶油在偏振光显微镜下的晶体微观形态。由图3可见,小烛树蜡凝胶油表现为簇状的晶体形态,结晶颗粒较大,随着温度的升高,晶体尺寸减小。日本木蜡凝胶油呈现出针状的晶体结构,结晶细小,随着温度的升高,晶体更容易发生部分聚集。此外,分形维数(Db)能够较好地定量分析结晶体系的均匀度[27]。从分形维数(Db)分析,小烛树蜡凝胶油比日本木蜡凝胶油具有更高的Db值,因而其具有更加稳定且有序的晶体结构,同时随着温度的升高,两种蜡基凝胶油的Db值均下降,说明两种凝胶油样品均在4℃具有更加致密而稳定的晶体结构。根据文献[28]报道,PLM下的针状晶体表现为具有较大比表面积的片层状晶体的边缘结构(冷冻扫描电子显微镜观察),这种晶体结构具有较强的油结合能力,这也解释了日本木蜡凝胶油在Db值较低的情况下仍然表现出强机械性能的原因。

2.3.2 晶型与晶体多态性
小烛树蜡和日本木蜡与其凝胶油的XRD图谱如图4所示。
由XRD获得的长、短间距信息通常用来表征脂肪晶体的层间排列结构和同质多晶现象[29]。由图4可知,在小角X射线衍射图中,纯蜡可以观察到长间距衍射峰,而凝胶油中没有,这与凝胶油中存在大量的液态甘油三酯(非晶态)有关。小烛树蜡在41.74 Å附近出现信号峰,这与之前的研究[19]类似,说明小烛树蜡存在长链正构烷烃,并以正交亚晶胞排列方式排布。日本木蜡则在42.13 Å和14.16 Å附近出现信号峰,由于日本木蜡的主要成分为甘油三酯,其纳米层状结构呈现二倍链长堆积结构。此外,小烛树蜡及小烛树蜡凝胶油的小角衍射峰(分别为21.34 Å和21.48 Å)均显示出比广角衍射峰更低的衍射强度,表明其晶体的层状平面内分子相互作用高于其C—C分子间相互作用。日本木蜡在14.16 Å的衍射峰则与广角衍射峰强度相似,说明日本木蜡可能存在较强的C—C分子间相互作用。由图4中广角X射线衍射图可知,小烛树蜡及小烛树蜡凝胶油在4.15 Å及3.7 Å附近出现2个衍射峰,表现为β′晶型。而日本木蜡则在4.6、4.2 Å和3.8 Å附近出现多个衍射峰,这与饱和甘油三酯的晶型十分相似,说明日本木蜡同时具有β和β′两种晶型,但在日本木蜡凝胶油中,仅在4.6 Å附近出现1个衍射峰,由此可知在日本木蜡凝胶油的结晶网络形成过程中,高油酸葵花籽油的加入使得脂肪晶体堆积排列方式改变,从而使日本木蜡晶型由β′型转为β型。
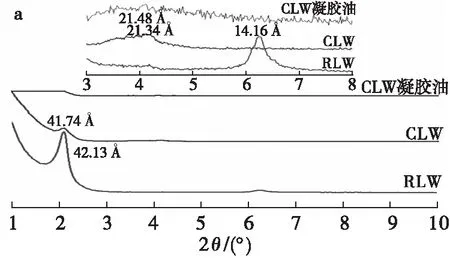
图4 小烛树蜡(CLW)和日本木蜡(RLW)及其凝胶油的XRD小角衍射图(a)和广角衍射图(b)
2.3.3 分子间作用力
为进一步探究两种蜡形成凝胶油的分子间作用力,采用傅里叶红外光谱仪(FT-IR)测定小烛树蜡、日本木蜡及其凝胶油和高油酸葵花籽油(HOSO)的红外谱图,结果如图5所示。

图5 小烛树蜡(CLW)、日本木蜡(RLW)及其凝胶油和高油酸葵花籽油(HOSO)的FT-IR谱图

3 结 论
本文系统地表征了小烛树蜡和日本木蜡两种不同熔点的蜡在高油酸葵花籽油中的结晶行为和网络结构差异。首先对比了两种蜡基凝胶油的流变学性质,二者均表现出低频率依赖性的“强凝胶”特征,高熔点的小烛树蜡比日本木蜡具有更强的热稳定性,小烛树蜡凝胶油具有较高的表观黏度,日本木蜡凝胶油则触变回复性更强。此外,两种蜡的热力学性质也存在较大差异,小烛树蜡具有更高的焓值,形成的网络结构更强,日本木蜡凝胶油结晶起始温度低,可在低温下结晶。在微观层面上,PLM结果显示小烛树蜡凝胶油和日本木蜡凝胶油分别呈簇状和针状晶体形态,二者的晶体尺寸和均匀度随温度的升高而下降,小烛树蜡比日本木蜡更易形成致密有序的晶体结构。在纳米层面上,小烛树蜡及其凝胶油的晶型始终为β′型,而日本木蜡则在形成结晶网络的过程中晶体堆积排列方式改变从而使晶型发生改变,由β′型转为更稳定的β型。分子间作用力证明在结晶网络形成过程中未出现氢键,蜡基凝胶油的网络结构中可能存在范德华力作用。日本木蜡具有迥异于其他高熔点蜡的结晶行为,为塑性脂肪替代提供了新的思路。
猜你喜欢
纺织学报(2022年3期)2022-03-28
人工晶体学报(2021年10期)2021-11-26
疯狂英语·新读写(2021年5期)2021-11-23
陶瓷学报(2021年1期)2021-04-13
发明与创新(2020年31期)2020-12-20
金桥(2020年10期)2020-11-26
军事文摘(2020年20期)2020-11-16
小猕猴学习画刊·下半月(2020年8期)2020-07-28
食品与生活(2016年5期)2016-05-23
