
SARM1 participates in axonal degeneration and mitochondrial dysfunction in prion disease
2022-03-09 07:22MengYuLaiJieLiXiXiZhangWeiWuZhiPingLiZhiXinSunMengYangZhaoDongMingYangDongDongWangWenLiDeMingZhaoXiangMeiZhouLiFengYang
中国神经再生研究(英文版) 2022年10期
Meng-Yu Lai, Jie Li, Xi-Xi Zhang, Wei Wu, Zhi-Ping Li, Zhi-Xin Sun, Meng-Yang Zhao,Dong-Ming Yang, Dong-Dong Wang, Wen Li, De-Ming Zhao, Xiang-Mei Zhou,Li-Feng Yang
Abstract Prion disease represents a group of fatal neurogenerative diseases in humans and animals that are associated with energy loss, axonal degeneration, and mitochondrial dysfunction. Axonal degeneration is an early hallmark of neurodegeneration and is triggered by SARM1. We found that depletion or dysfunctional mutation of SARM1 protected against NAD+ loss, axonal degeneration, and mitochondrial functional disorder induced by the neurotoxic peptide PrP106-126. NAD+supplementation rescued prion-triggered axonal degeneration and mitochondrial dysfunction and SARM1 overexpression suppressed this protective effect.NAD+ supplementation in PrP106-126-incubated N2a cells, SARM1 depletion, and SARM1 dysfunctional mutation each blocked neuronal apoptosis and increased cell survival. Our results indicate that the axonal degeneration and mitochondrial dysfunction triggered by PrP106-126 are partially dependent on SARM1 NADase activity. This pathway has potential as a therapeutic target in the early stages of prion disease.
Key Words: axonal degeneration; mitochondrial dysfunction; NAD+ metabolism; NADase; neurodegenerative disease; prion disease; SARM1; sterile alpha and TIR motif-containing 1
Introduction
Prion disease represents a group of transmissible and fatal neurogenerative diseases in both animals and humans that includes scrapie, bovine spongiform encephalopathy, and Creutzfeldt-Jakob disease (Collinge, 2001). Accumulation of the misfolded form of the cellular prion protein in the central nervous system induces neuronal loss, spongiform vacuolation, and inflammation and is the main cause of neurodegeneration (Prusiner, 1991; Hur et al., 2002;Soto and Satani, 2011; Batlle and Ventura, 2020). The misfolded cellular prion protein is termed PrPSc. Prion protein peptide 106-126 (PrP106-126), a neurotoxic prion protein fragment, has been used by researchers to mimic amyloid-like fibril accumulation and trigger axonal degeneration and neuronal apoptosisin vitro(Prusiner, 1991; Soto and Satani, 2011; Forloni et al., 2019;Groveman et al., 2020; Ishikawa et al., 2021).
In studies of various neurodegenerative diseases, the loss of synapses and axons that precedes neuronal loss is adequate to cause clinical symptoms(Li et al., 2001; Gunawardena and Goldstein, 2005; Luo and O’Leary,2005; Stokin and Goldstein, 2006; Gerdts et al., 2015). Therefore, axonal degeneration is considered a hallmark of neurodegenerative disorders(Soto and Satani, 2011). Mitochondria are the most important cellular hub for adenosine triphosphate (ATP) production, the key source of cellular energy. Neurons are especially sensitive to mitochondrial dysfunction due to their extreme dependence on mitochondria for energy (Galluzzi et al., 2012; Morán et al., 2012). During ATP biosynthesis, nicotinamide adenine dinucleotide (NAD+) functions as an irreplaceable coenzyme. In previous studies, researchers found that the fifth member of the myeloid differentiation primary response 88 family, sterile alpha and Toll-interleukin receptor 1 receptor (TIR) motif-containing 1 (SARM1), was required for axonal degeneration induced by axotomy. In addition, SARM1 knockout rescued symptoms in bothin vivoandin vitromodels (Osterloh et al., 2012;Henninger et al., 2016). Because injury-induced axonal degeneration has been associated with reduced NAD+concentration, SARM1 may regulate this degeneration via a gain of NAD+cleavage activity following release of SARM1 autoinhibition and dimerization of its TIF domains (Wang et al., 2005; Gerdts et al., 2015).
Neuronal death and axonal degeneration in prion disease are linked with NAD+depletion (Wang et al., 2015) and can be relieved by NAD+replenishment. In a previous study, we investigated prion disease-induced mitochondrial dysfunction in both cell line and animal models (Wu et al.,2019). Because mitochondria process NAD+to synthesize ATP and SARM1 can link to the outer mitochondrial membrane via its mitochondrial targeting sequence domain (Panneerselvam et al., 2012), we hypothesized that axonal degeneration associated with prion disease is partially regulated by SARM1 protein activation and its subsequent destruction of NAD+. To verify this hypothesis, we examined the effect of SARM1 knockdown/null mutation on the NAD+depletion, mitochondrial dysfunction, and axonal degeneration induced by treatment with PrP106-126. In addition, we determined the effect of NAD+replenishment on mitochondrial dysfunction and mitochondrial and axonal morphology in cells incubated with PrP106-126.
Materials and Methods
Ethics statement
All animals in this study were treated in accordance with the Guidelines of the China Agricultural University on the Review of Welfare and Ethics of Laboratory Animals. The study protocol was approved by the China Agricultural University Administration Office of Laboratory Animals (approval No. AW02011202-2-1) on September 1, 2020.
Cell culture and treatment
Primary neurons were prepared from the cerebral cortex of 1-day-old neonatal Sprague-Dawley rats (SPF (Beijing) Biotechnology Co., Ltd. Beijing, China;license No. SCXK (Jing) 2016-0002) following a previously described procedure(Song et al., 2016). Neonatal rats were sacrificed by decapitation. Neurons were dissociated from cerebral cortex tissue after digestion by using Dulbecco’s modified Eagle’s medium (Hyclone, Logan, UT, USA) with papain (2 mg/mL,Invitrogen, Carlsbad, CA, USA) and DNAse (50 mg/mL; Sigma-Aldrich, St. Louis,MO, USA) for 30 minutes at 37°C. The cells were plated and pre-incubated with L-polylysine (Solarbio, Beijing, China) at the appropriate concentration for different uses. Primary cortical neurons were cultured in a specific culture medium comprising Neurobasal-A Medium (Invitrogen) with 2% serum-free B27 supplement (Invitrogen) and 0.5% penicillin-streptomycin (Gibco, Grand Island, NY, USA). The medium was replaced every 2 days in half of the volume to satisfy the nutritional requirements for cell growth. The experimental processes were performed after 1 week of culture. Culture medium used for mouse neuroblastoma (N2a) cells (National Infrastructure of Cell Line Resource, Beijing, China; ATCC Cat# CCL-131, RRID: CVCL_0470) was based on Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum(Gibco) and 1% penicillin-streptomycin (Gibco). Primary neurons and N2a cells were cultured at 37°C in a humid incubator with 5% CO2.
PrP peptide PrP106-126(sequence: KTNMKKHMAGAAAAGAVVGGLG; 98% purity;Sangon Bio-Tech, Beijing, China) was dissolved in phosphate-buffered saline(PBS; Solarbio) to form a 1 mM storage solution. After being shaken at 4°C for 24 hours, the solution could be stored at 4°C for a short period. Primary neurons or N2a cells were incubated with the peptide for 3, 6, 12, or 24 hours as described in the results. The final peptide concentration in experiment processing was 200 μM.
β-Nicotinamide adenine dinucleotide (β-NAD; Sigma-Aldrich) was dissolved in PBS to a storage concentration of 15 μm/mL and stored away from light at-20°C. Primary neurons or N2a cells were incubated with β-NAD for 12 hours with PrP peptide.
Small interfering RNA, plasmids, and transfection
The SARM1 small interfering RNA (siRNA; 5′-GGT AGA TGG TGA TTT GCT T-3′),pCMV-SARM1 plasmid, and pCMV-SARM1(S548A) plasmid were obtained from SYKMGENE (Beijing, China). Primary neurons were transfected with antibioticfree Dulbecco’s modified Eagle’s medium containing 500 ng siRNA, 0.75 μL Lipofectamine 3000, and 50 μL Opti-MEM (both from Invitrogen) per 500 μL cell culture medium for 48 hours. N2a cells were transfected with siRNA using the same reagent system for 48 hours or with siRNA for 12 hours as a protein knockdown background and then transfected with the SAMR1 plasmid or SARM1(S548A) plasmid using the same reagent system but with adding 1 μL P3000 reagent (Invitrogen) per 500 μL culture medium for 48 hours.
Western blot assay
Both primary neurons and N2a cells were harvested after the experimental processes using lysis buffer (Beyotime, Shanghai, China) containing 1%protease inhibitor solution (Beyotime). The lysed cells were collected and cultured with lysis buffer in Eppendorf tubes and centrifuged at 12,000 × g for 10 minutes at 4°C. The supernatants were then transferred to new tubes.The samples were used for western blotting after boiling with loading buffer(Solarbio).
Proteins were separated on sodium dodecyl sulfate-polyacrylamide gels according to molecular weight and then transferred onto a nitrocellulose membrane (Sigma-Aldrich). After culturing with 5% skim milk dissolved in Tris-buffered saline containing 0.1% Tween 20, the samples were placed in blocking buffer at 37°C for 1 hour. Individual primary antibodies were incubated at appropriate concentrations at 4°C overnight, including rabbit anti-SARM1 (Abcam, Cambridge, UK, Cat# ab226930, RRID: AB_2893433,1:1000), mouse anti-neuronal class III tubulin (TU20; CST, Danvers, MA, USA,Cat# 4466, RRID: AB_1904176, 1:1000), mouse anti-neuronal class III tubulin(Tuj1; Beyotime, Cat# AT809, RRID: AB_2893434, 1:500), and mouse antiglyceraldehyde-3-phosphate dehydrogenase (Proteintech, Rosemont, IL, USA,Cat# 60004-1-Ig, RRID: AB_2107436, 1:10,000). After each membrane was washed with Tris-buffered saline containing 0.1% Tween 20, it was incubated with relevant secondary antibodies, including goat anti-mouse IgG conjugated to horseradish peroxidase (ZSGB-Bio, Beijing, China, Cat# ZB2305, RRID:AB_2747415, 1:10,000) and goat anti-rabbit IgG conjugated to horseradish peroxidase ZSGB-Bio, Cat# ZB2301, RRID: AB_2747412, 1:10,000). The membranes were imaged using a gel imaging system (Tanon, Shanghai, China)and results were normalized to glyceraldehyde-3-phosphate dehydrogenase.Images were analyzed using ImageJ software version 1.52a (National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012).
Immunofluorescence staining
Primary neurons were seeded on 24-well cell culture plates with cover slips pre-incubated with L-polylysine (Solarbio) after the dissociation steps described above. After experimental treatment, cells were softly washed with PBS and bathed with Immunolo Staining Fix Solution (Beyotime), followed by Immunol Permeabilization Buffer (Beyotime) at room temperature for 10 minutes, and Immunol Staining Blocking Buffer (Beyotime) at room temperature for 1 hour. Primary antibody incubations were processed at 4°C overnight followed by relevant secondary antibody incubations at 37°C for 1 hour. Primary neuronal axons were labelled using a mouse anti-TU20 antibody(a marker of primary neuronal axons; CST, Cat# 4466, RRID: AB_1904176,1:1000) and an Alexa Fluor 488 labeled goat anti-mouse IgG (H+L) (Beyotime,Cat# A0428, RRID: AB_2893435, 1:400). Mitochondria in primary neurons were labelled using rabbit anti-translocase of outer mitochondrial membrane 40 (TOMM40) antibody (Proteintech, Cat# 18409-1-AP, RRID: AB_2303725,1:1000) and Alexa Fluor 555-labeled donkey anti-rabbit IgG (H+L) antibody(Beyotime, Cat# A0453, RRID: AB_2890132, 1:400). All slides were photographed using a Nikon (Tokyo, Japan) confocal imaging system. Images were analyzed using ImageJ software version 1.52a.
Determination of mitochondrial function
The JC-1 mitochondrial membrane potential (MMP) assay kit (Beyotime) was used to detect the MMP. The N2a cells were harvested after experimental processes, collected into a tube after washing softly by PBS, and then incubated with the JC-1 probe for 30 minutes at 37°C. After washing with the wash buffer from the kit, the samples were analyzed using flow cytometry.The ATP determination kit (Beyotime) was used to measure cellular ATP level, which was performed at low temperature. Cells in the culture plate were lysed with the lysis buffer from the kit and incubated in black 96-well plates with 100 μL detection reagent; 50 μL samples were added to each well. Luminescence of each well was measured using a luminometer (TECAN,Männedorf, Austria).
Measurement of NAD level
The NAD/NADH-Glo assay kit (Promega, Madison, WI, USA) was used to determine cellular NAD. After experimental processes, N2a cells were reseeded at a concentration of 30,000 cells/well in flat white 96-well plates and incubated with detection reagent preformed as guidance of manufacturer instructions for 30 minutes. Luminescence was recorded using a plate stacker(TECAN).
Cell viability assay
The Cell Counting Kit-8 assay kit (Beyotime) was used to determine N2a cell viability after experimental processes. The kit detection reagent was diluted with cell culture medium according to manufacturer instructions and incubated with the cells for 1 hour under cell culture conditions in a humid incubator. Using a cell-free sample as blank control, the absorbance at 450 nm was determined using a microplate reader (Thermo, Waltham, MA, USA). Cell viability was expressed as a percentage of the untreated control sample (100%).
TdT-mediated dUTP nick-end labeling assay
N2a cells were seeded on cover slips in a 24-well plate. After the experimental treatments, the One Step TdT-mediated dUTP nick-end labeling (TUNEL)Apoptosis Assay Kit (Beyotime) was used. This procedure was performed away from light. The samples were prepared as described for immunofluorescence microscopy until antibody incubation. Then, the samples were incubated with TUNEL stain reagent for 1 hour at 37°C. 4′,6-Diamidino-2-phenylindole stain reagent (Beyotime) was used to visualize the nucleus for 10 minutes at 37°C.All slides were photographed using a Nikon confocal imaging system. Images were analyzed using ImageJ software version 1.52a.
Statistical analysis
Data are expressed as means ± standard error of the mean (SEM) and were analyzed using one-way analysis of variance followed by Bonferroni’spost hoctest. Analyses were performed using Prism 5.0 software (GraphPad Software,La Jolla, CA, USA).P< 0.05 was considered significant.
Results
Axonal degeneration induced by PrP106-126 incubation
In prion disease, the most characteristic pathological change is neuronal degeneration, which causes dramatic clinical symptoms (Prusiner, 1991; Hur et al., 2002; Soto and Satani, 2011). Neuronal cell damage in prion disease models has been investigated in our previous studies (Wang et al., 2015; Song et al., 2016), which focused on morphological changes in the ultrastructure of primary cultured cortical neurons and axonal loss by spinal cord neurons after PrP106-126incubation. We found that the axons of primary cultured cortical neurons were severely shortened after 12 hours of incubation with 200 μM PrP106-126peptide (Figure 1A and B). In N2a cells, the expression level of beta-tubulin III, a neuron-specific type of tubulin (Gerdts et al., 2013), was reduced remarkably by the same treatment (Figure 1C and D). These results demonstrate that 12 hours of incubation with PrP106-126 caused axonal degeneration in primary cultured cortical neurons and decreased beta-tubulin III expression in N2a cells.
SARM1 is required for PrP106-126-induced axonal degeneration
In recent studies of neurodegenerative diseases, the role of SARM1 in axonal degeneration has gradually become clearer (Osterloh et al., 2012; Gerdts et al., 2013; Henninger et al., 2016; Essuman et al., 2017). To determine whether SARM1 is involved in axonal degeneration induced by PrP106-126incubation,we first examined the level of SARM1 expression in N2a cells. According to our western blot results, SARM1 expression was increased significantly in N2a cells treated with PrP106-126for 12 or 24 hours (Figure 2A and B). Since SARM1-expressing neurons showed decreased expression of beta-tubulin III after treatment with PrP106-126, we explored the role of SARM1 in this effect with SARM1 knockdown using siRNA (Figure 2C). Western blotting showed that SARM1 knockdown N2a cells maintained their level of beta-tubulin III following treatment with PrP106-126(Figure 2D and E). Next, we overexpressed full-length SARM1 using plasmid transfection (500 ng for 48 hours) in SARM1 knockdown N2a cells (Figure 2F) and showed that treatment with PrP106-126reduced the beta-tubulin III expression level of the transfected cells (Figure 2G and H). In addition, SARM1 knockdown (Figure 2I) protected the axonal structure of primary cultured cortical neurons during PrP106-126incubation(Figure 2J and K). Our findings indicate that SARM1 is involved in axonal degeneration caused by PrP106-126and that knockdown of SARM1 protects neurons from this pathological change.
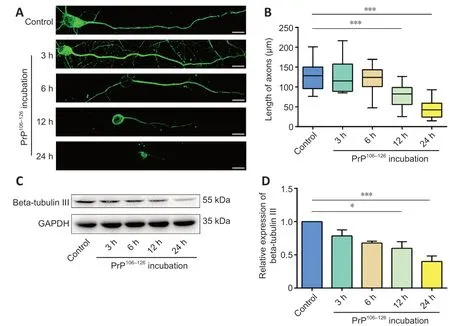
Figure 1 |PrP106-126 triggers axonal degeneration in primary cultured neurons and N2a cells.(A) Rat primary cultured neurons were incubated with 200 μM PrP106-126 for 3, 6, 12, or 24 hours. Neuronal structure was photographed after immunofluorescence staining with anti-TU20 (green, stained by Alexa Fluor 488). Scale bars: 10 μM. Axon length declined significantly after incubation with 200 μM PrP106-126 for 12 and 24 hours. (B) Analysis of axon length after PrP106-126 incubation. (C) Western blot bands of beta-tubulin III in PrP106-126-treated N2a cells. (D) Quantitative results of tubulin III expression, which was normalized to the control group. Expression of beta-tubulin III declined significantly after incubation with 200 μM PrP106-126 for 12 and 24 hours. Data are expressed as means ± SEM. All assays were repeated three times. *P < 0.05, ***P < 0.001, vs. control group (one-way analysis of variance followed by Bonferroni’s post hoc test). GADPH:Glyceraldehyde-3-phosphate dehydrogenase; PrP106-126: PrP peptide 106-126; TU20:neuronal class III tubulin.
PrP106-126-induced mitochondrial morphological damage and dysfunction
are partly mediated by SARM1
Considering that neurons consume a large amount of energy, mitochondria are critical organelles involved in many neurodegenerative diseases (Pacelli et al., 2015; Swerdlow, 2018; Wang et al., 2021). In our previous studies,mitochondrial fragmentation and dysfunction were observed in prion disease models (Li et al., 2018; Wu et al., 2019; Zhang et al., 2020). In other studies,SARM1 function was also associated with mitochondrial respiration and mitochondrial dysfunction-induced cell death (Summers et al., 2014; Murata et al., 2018). Therefore, we hypothesized that SARM1 plays a role in PrP106-126-induced axonal degeneration via regulation of mitochondrial function. After incubation with 200 μM PrP106-126for 12 hours, mitochondria in primary cultured cortical neurons were imaged using fluorescence microscopy.TOMM40 staining showed that PrP106-126-treated neurons had a remarkably fragmented mitochondrial network, whereas untreated control neurons had filamentous, continuous mitochondria, which suggested mitochondrial changes. The mitochondria of primary neurons that were treated with SARM1 siRNA and then incubated with PrP106-126maintained their tubular morphology(Figure 3A). Mitochondrial length decreased significantly in PrP106-126-treated neurons; however, in PrP106-126-treated SARM1-knockdown neurons,the length significantly recovered (Figure 3B). To ascertain mitochondrial function, we measured the ATP level of PrP106-126-treated N2a cells. After PrP106-126incubation, the ATP level of N2a cells was reduced to approximately 50% of that of control. PrP106-126-induced ATP loss was prevented by SARM1 knockdown and accelerated by overexpression of SARM1 (Figure 3C).
MMP loss is also a signal of mitochondrial dysfunction; therefore, we assessed the MMP using the JC-1 assay. The monomer form of JC-1 in damaged mitochondria appears green, while the aggregate form in healthy mitochondria appears red. The red/green ratio reflects the MMP and would decrease with MMP loss. In concert with the ATP measurements mentioned above, PrP106-126incubation lowered the MMP compared with the untreated control. The change in MMP induced by PrP106-126incubation was prevented by SARM1 knockdown and aggravated by SARM1 overexpression (Figure 3D).These results indicate that SARM1 takes part in the mitochondrial morphology changes and dysfunction induced by PrP106-126incubation.
PrP106-126-induced mitochondrial dysfunction and axon degeneration via the NAD+-hydrolase activity of SARM1
In a previous study, Wang et al. (2015) proposed NAD+replenishment for neuroprotection in patients with prion disease. Because SARM1 performs cleavage of NAD+, which has been associated with axonal degeneration (Gerdts et al., 2015; Essuman et al., 2017), we conjectured that SARM1 mediates axonal degeneration and mitochondrial dysfunction via its NAD+hydrolase activity. We found that PrP106-126incubation destroys approximately 50%of cellular NAD+. In cells incubated with PrP106-126and treated with SARM1 siRNA, the NAD+level was similar to that in control cells; however, cells overexpressing SARM1 showed enhanced loss of NAD+(Figure 4A). Next, we investigated the effects of NAD+supplementation on axonal and mitochondrial morphology in anin vitroprion model by observing β-NAD-treated primary neurons using fluorescence microscopy. In primary cultured cortical neurons incubated with PrP106-126, the mitochondria of β-NAD-treated cells maintained their filamentous and tubular shape and longer average length compared with the fragmented mitochondria of the PrP106-126-treated group (Figure 4B and C). ATP level and MMP were measured to assess mitochondrial function.The ATP level in PrP106-126-incubated N2a cells was increased dramatically after treatment with β-NAD. Therefore, NAD treatment protected cells from PrP106-126-induced ATP loss, even in cells overexpressing SARM1. However,SARM1 overexpression impaired recovery of the ATP level (Figure 4D).
In concert with the findings described above, after PrP106-126incubation,the MMP of NAD-treated cells was maintained compared with that of cells treated with PrP106-126only. Moreover, overexpression of SARM1 weakened the protective effect of NAD supplementation on MMP during PrP106-126incubation (Figure 4E). In cells incubated with PrP106-126, axons in the β-NADtreated group were significantly longer (Figure 4B and F). NAD+treatment also resulted in higher expression of beta-tubulin III in N2a cells; however, and not surprisingly, overexpression of SARM1 limited the recovery of beta-tubulin III expression (Figure 4G and H).
Our results suggest that the NAD+hydrolase activity of SARM1 plays a role in PrP106-126-induced mitochondrial dysfunction and axonal degeneration, and that NAD supplementation can prevent these deleterious effects of PrP106-126on mitochondria and axons.
The reduced NADase activity of the SARM1(S548A) mutant limits mitochondrial dysfunction and axonal degeneration triggered by PrP106-126
To confirm that the NAD+hydrolase activity of SARM1 is a critical factor in mitochondrial dysfunction and axonal degeneration, we obtained a plasmid containing the S548A mutant SARM1 sequence, as S548A SARM1 has reduced NAD+cleavage activity (Murata et al., 2018). After transfecting the SARM1(S548A) plasmid into SARM1-silenced N2a cells (Figure 5A), we measured the intracellular NAD+level in cells treated with PrP106-126and cells that were not treated. NAD+depletion induced by PrP106-126incubation in the SARM1(S548A)-expressed N2a cells was limited and similar to that in the SARM1-silenced group but dramatically higher than that in the SARM1-overexpressed group (Figure 5B). Next, we measured the mitochondrial function of SARM1(S548A) N2a cells (Figure 5C and D). In contrast to cells overexpressing SARM1, SARM1(S548A) N2a cells did not show a loss of ATP or reduced MMP following PrP106-126incubation. The intracellular ATP concentration and MMP of SARM1(S548A) N2a cells were maintained at levels similar to those in the SARM1-silenced group. Analysis of beta-tubulin III expression via western blotting yielded similar results (Figure 5E and F).Incubation with PrP106-126did not decrease the expression of beta-tubulin III in SARM1(S548A) N2a cells. These results suggest that the NAD-hydrolase activity of SARM1 is required for SARM1-related mitochondrial dysfunction and axonal degeneration induced by PrP106-126incubation.
SARM1 is involved in PrP106-126-induced neuronal death
Because neuronal apoptosis is a characteristic of prion disease (Forloni et al.,2019; Wu et al., 2019) and cell survival is a key clinicopathologic factor, we investigated the effect of SARM1 NAD+hydrolase activity in cells incubated with PrP106-126. Apoptosis was assessed using the TUNEL assay in control,SARM1-silenced, SARM1-overexpressed, and SARM1(S548A)-expressed N2a cells (Figure 6A and B). The apoptotic cell percentage was increased in N2a cells following PrP106-126incubation. This increase was prevented in SARM1-silenced cells and enhanced in SARM1-overexpressed cells. In the SARM1(S548A)-expressed group, PrP106-126incubation failed to trigger apoptosis. Next, we assayed cell viability using the CCK8 assay (Figure 6C).SARM1 silencing protected PrP106-126-incubated cells from dying, while SARM1 overexpression increased the percentage of apoptotic cells. In addition,cell viability was maintained in SARM1(S548A)-expressed cells treated with PrP106-126, confirming the results of the apoptosis assays. These results indicate that the NAD+hydrolase activity of SARM1 is critical in PrP106-126-induced cell death and that SARM1 silencing or loss of function protects against this PrP106-126effect.
Pharmacological supplementation with NAD+ promotes N2a survival in
prion disease
As shown in Figure 4, NAD+supplementation protected mitochondrial morphology and function from the deleterious effects of PrP106-126; therefore,we assessed the effect of NAD+supplementation on cell survival. The results of apoptosis and viability assays showed that NAD+supplementation inhibited apoptosis induced by PrP106-126and increased cell survival. In the SARM1-overexpressed group, NAD+supplementation inhibited apoptosis and protected cells from cell death induced by PrP106-126; but the protective effect was limited by SARM1 overexpression (Figure 7A-C). These results confirm that NAD+supplementation rescued PrP106-126-induced apoptosis and that this effect was suppressed by SARM1 overexpression.
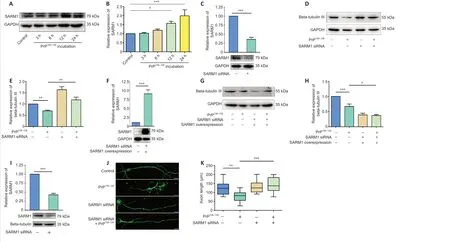
Figure 2 | SARM1 contributes to axon degeneration in PrP106-126-treated cells.(A) Western blots of SARM1 in PrP106-126-treated N2a cells. (B) Quantitative results for SARM1 expression. (C) Representative western blot results for SARM1 protein in N2a cells after transfection with SARM1 siRNA for 48 hours. (D) Western blots of beta-tubulin III in SARM1 knockdown N2a cells treated with PrP106-126 compared with control. (E) Quantitative results for the data shown in D. (F) Representative western blot results for SARM1 protein in N2a cells transfected with SARM1 plasmid for 48 hours after 12 hours SARM1 siRNA transfection. (G)Western blots of beta-tubulin III in SARM1 overexpressed N2a cells treated with PrP106-126 compared with control. (H) Quantitative results for the data shown in G. (I) Representative western blot results for SARM1 protein in primary neurons after transfection with SARM1 siRNA for 48 hours. (J) Primary neurons were transfected with SARM1 siRNA for 48 hours before incubation with PrP106-126. Neuronal structure is shown in fluorescent green (stained by Alexa Fluor 488). After incubation with PrP106-126, axonal length was shortened significantly. SARM1 knockdown protected axons from PrP106-126-induced degeneration. Scale bars: 10 μm. (K) Analysis of axon length. Data are expressed as the mean ± SEM. All assays were repeated three times. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni’s post hoc test). GADPH: Glyceraldehyde-3-phosphate dehydrogenase; PrP106-126: PrP peptide 106-126; SARM1: sterile alpha and Toll-like receptor motif-containing 1; siRNA: small interfering RNA.
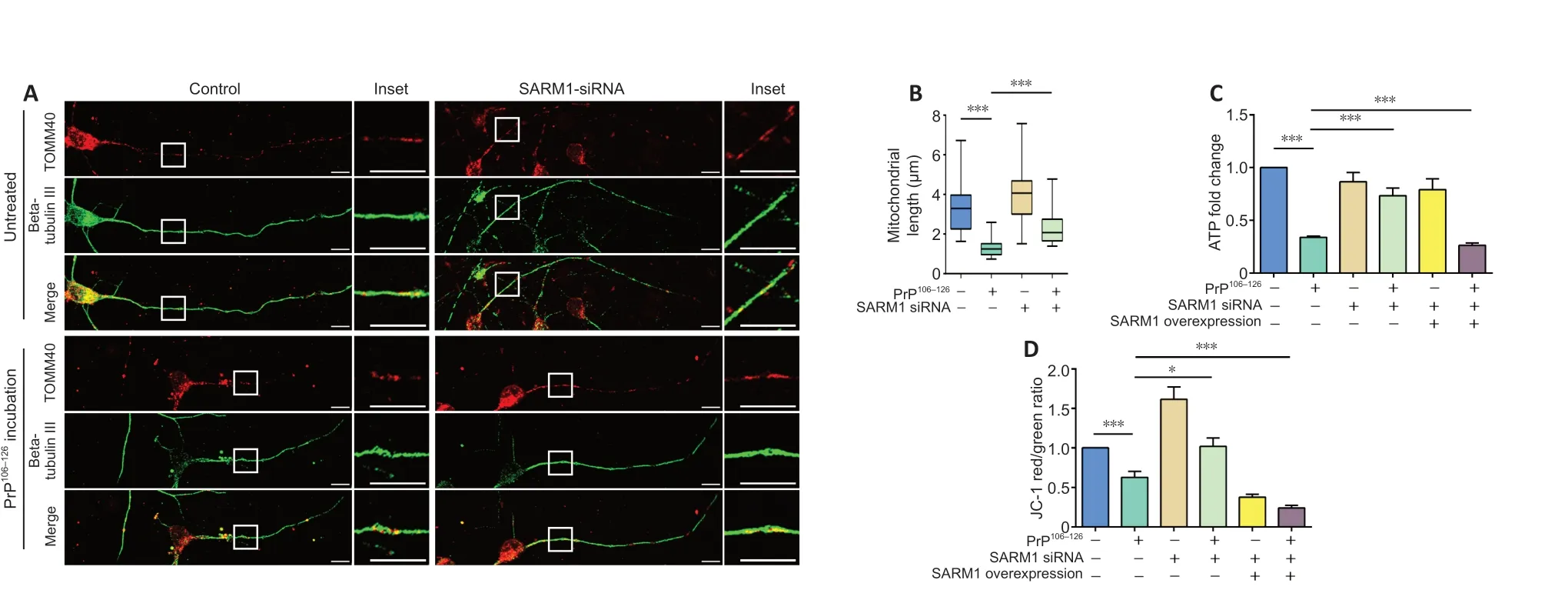
Figure 3 | SARM1 is involved in changes in mitochondrial morphology and function caused by PrP106-126 treatment.(A) Primary neurons were transfected with SARM1 siRNA for 48 hours before PrP106-126 incubation. After incubation with PrP106-126, the mitochondrial network was fragmented;however, in the SARM1 knockdown group, mitochondria kept their tubular morphology after PrP106-126 incubation. Neuronal structure is shown in fluorescent green (Alexa Fluor 488),while mitochondria appear red (Alexa Fluor 555). Scale bars: 10 μM. (B) Analysis of mitochondrial length. (C) Relative intracellular ATP level. Control, SARM1-silenced, and SARM1-overexpressed N2a cells were incubated with PrP106-126. (D) Mitochondrial membrane potential. Control, SARM1-silenced, and SARM1-overexpressed N2a cells were incubated with PrP106-126. Data are expressed as means ± SEM. All assays were repeated three times. *P < 0.05, ***P < 0.001 (one-way analysis of variance followed by Bonferroni’s post hoc test). ATP:Adenosine triphosphate; PrP106-126: PrP peptide 106-126; SARM1: sterile alpha and Toll-like receptor motif-containing 1; siRNA: small interfering RNA; TOMM40: translocase of outer mitochondrial membrane 40.
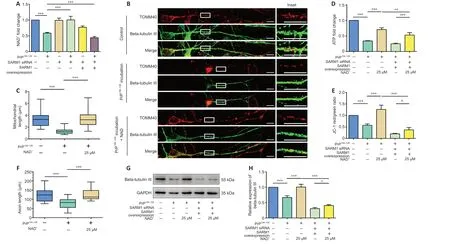
Figure 4 | NAD-hydrolase activity of SARM1 plays a role in PrP106-126-induced axonal degeneration and mitochondrial disorders.(A) Relative intracellular NAD level. Control, SARM1-silenced, and SARM1-overexpressed N2a cells were incubated with or without PrP106-126. (B) Primary neurons were incubated with 25 μM NAD and 200 μM PrP106-126 for 12 hours. After PrP106-126 incubation, the intracellular NAD level dropped significantly; however, in SARM1-silenced cells, the NAD level was protected. SARM1-overexpression enhanced the decrease in intracellular NAD. Neuronal structure is shown in fluorescent green (Alexa Fluor 488), while mitochondria appear red (Alexa Fluor 555). Scale bars: 10 μm. (C) Analysis of mitochondrial length. (D) Relative intracellular ATP levels in control and SARM1-overexpressed N2a cells incubated with PrP106-126 with and without NAD treatment. (E) Mitochondrial membrane potential in control and SARM1-overexpressed N2a cells incubated with PrP106-126 with and without NAD treatment. (F) Analysis of axon length. (G) Western blots of beta-tubulin III levels in control and SARM1-overexpressed N2a cells incubated with PrP106-126 with and without NAD treatment. (H) Quantitative results of the data shown in G. Data are expressed as means ± SEM. All assays were repeated three times. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni’s post hoc test). GADPH: Glyceraldehyde-3-phosphate dehydrogenase; PrP106-126: PrP peptide 106-126; NAD: nicotinamide adenine dinucleotide; SARM1: sterile alpha and Toll-like receptor motif-containing 1; siRNA: small interfering RNA; TOMM40: translocase of outer mitochondrial membrane 40.

Figure 5 | Expression of SARM1(S548A) improves mitochondrial dysfunction and reduces axonal degeneration induced by PrP106-126.(A) Western blot results for SARM1 protein in N2a cells transfected with the SARM1(S548A) plasmid for 48 hours after 12 hours SARM1 siRNA transfection. (B) Relative intracellular NAD level. Control, SARM1-silenced, SARM1-overexpressed, and SARM1(S548A)-expressed N2a cells were incubated with or without PrP106-126. (C) Relative intracellular ATP levels in control, SARM1-silenced, SARM1-overexpressed, and SARM1(S548A)-expressed N2a cells incubated with or without PrP106-126. (D) Mitochondrial membrane potential in control,SARM1-silenced, SARM1-overexpressed, and SARM1(S548A)-expressed N2a cells incubated with or without PrP106-126. (E) Western blots of beta 3-tubulin in control, SARM1-silenced,SARM1-overexpressed, and SARM1(S548A)-expressed N2a cells incubated with or without PrP106-126. (F) Quantitative results of the data shown in E. Data are expressed as means ±SEM. All assays were repeated three times. **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni’s post hoc test). ATP: Adenosine triphosphate; GADPH:glyceraldehyde-3-phosphate dehydrogenase; NAD: nicotinamide adenine dinucleotide; ns: not significant; PrP106-126: PrP peptide 106-126; SARM1: sterile alpha and Toll-like receptor motif-containing 1; siRNA: small interfering RNA.
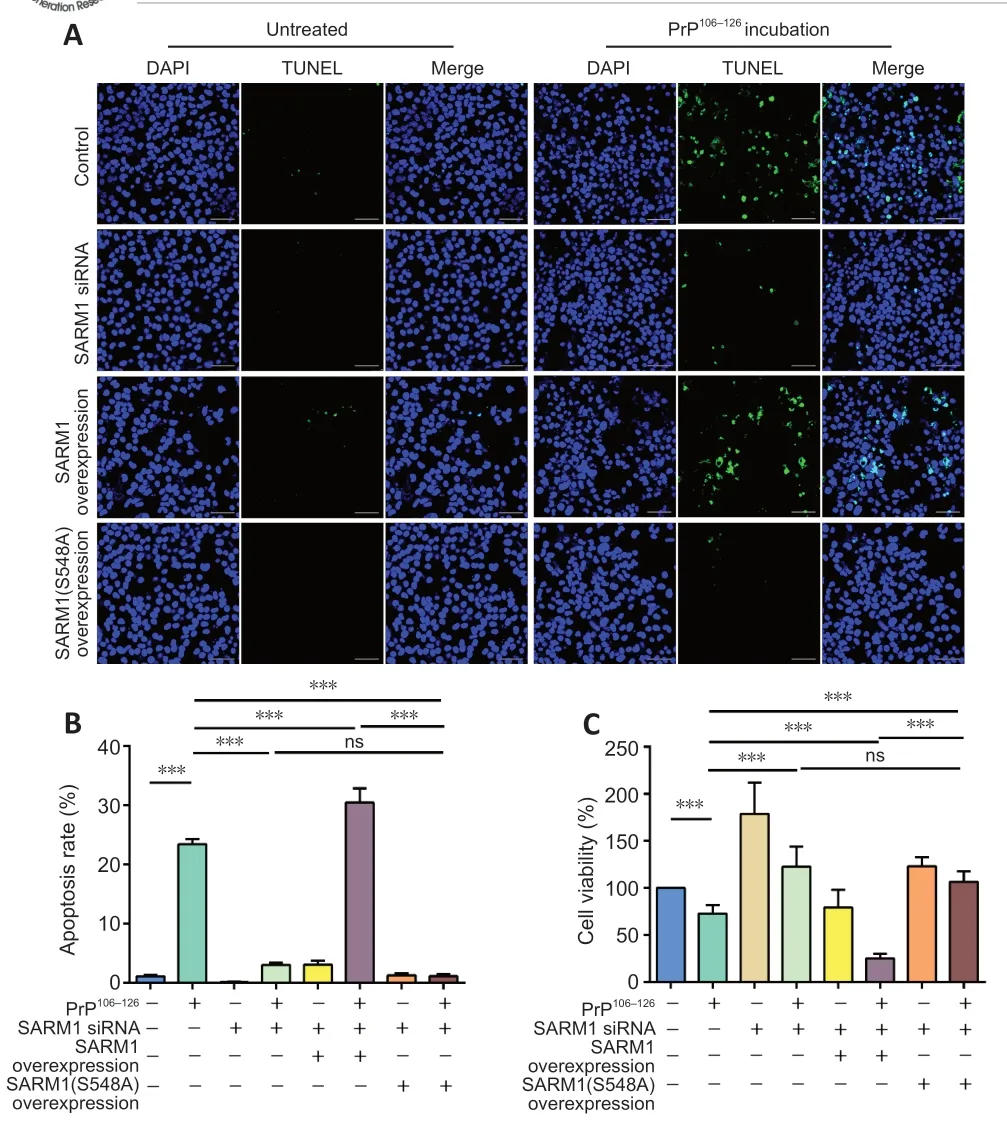
Figure 6 |NADase activity of SARM1 is associated with cell survival following incubation with PrP106-126.(A) TUNEL staining results in control, SARM1-silenced, SARM1-overexpressed, and SARM1(S548A)-expressed N2a cells incubated with or without PrP106-126. Incubation with PrP106-126 resulted in increased TUNEL stain intensity. SARM1-overexpression enhanced this increase while SARM1-silencing and SARM1(S548A) expression decreased it. Scale bars: 50 μm. (B) Analysis of apoptosis rate for the data shown in A. (C) N2a cell viability was assayed using the cell counting kit-8 kit in control, SARM1-silenced,SARM1-overexpressed, and SARM1(S548A)-expressed cells incubated with or without PrP106-126. Data are expressed as means ± SEM. All assays were repeated three times.***P < 0.001 (one-way analysis of variance followed by Bonferroni’s post hoc test).DAPI: 4′,6-Diamidino-2-phenylindole; NAD: nicotinamide adenine dinucleotide; ns: not significant; PrP106-126: PrP peptide 106-126; SARM1: sterile alpha and Toll-like receptor motif-containing 1; siRNA: small interfering RNA; TUNEL: TdT-mediated dUTP nick-end labeling.
Discussion
Prion disease represents a group of fatal central nervous system diseases that exhibit typical features of neurodegeneration, spongiform vacuolation,and inflammation (Soto and Satani, 2011). The pathogenesis of prion disease remains unclear, but axonal degeneration, mitochondrial dysfunction, and neuronal death have been described and neuronal loss is thought to be the cause of clinical symptoms and death (Hur et al., 2002; Song et al., 2016;Forloni et al., 2019). Evidence from the last decade suggests that SARM1 is associated with axonal degeneration in various pathologic conditions, such as injury, mitochondrial dysfunction, and Leber congenial amaurosis (Osterloh et al., 2012; Gerdts et al., 2013; Summers et al., 2014; Ko et al., 2020; Sasaki et al., 2020). We demonstrated that SARM1 knockdown N2a cells maintained a higher level of axon formation-associated beta-tubulin III expression following PrP106-126incubation. In addition, re-establishing expression of SARM1 led to PrP106-126-induced beta-tubulin III loss. SARM1 depletion also protected the axons of primary cultured cortical neurons from the deleterious effects of PrP106-126. This finding is in accordance with the studies mentioned above that used other disease models.

Figure 7 |NAD supplementation is protective against the deleterious effects of PrP106-126 incubation.(A) TUNEL staining results of control and SARM1-overexpressed N2a cells incubated with and without PrP106-126 and with and without NAD treatment. Incubation with PrP106-126 resulted in increased TUNEL stain intensity. In the condition of PrP106-126 incubation, NAD+supplementation decreased the stain intensity; SARM1 overexpression limited the effect of NAD+ supplementation. Scale bars: 50 μm. (B) Analysis of apoptosis rate for the data shown in A. (C) N2a cell viability was assayed using the cell counting kit-8 kit with control and SARM1-overexpressed N2a cells incubated with PrP106-126 with or without NAD treatment. Data are expressed as means ± SEM. All assays were repeated three times.**P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni’s post hoc test). DAPI: 4′,6-Diamidino-2-phenylindole; NAD: nicotinamide adenine dinucleotide;PrP106-126: PrP peptide 106-126; SARM1: sterile alpha and Toll-like receptor motifcontaining 1; siRNA: small interfering RNA; TUNEL: TdT-mediated dUTP nick-end labelling.
Fragmentation of axons and mitochondria after PrP106-126incubation is an indicator of neuronal dysfunction. As the main producer of energy in cells,mitochondria are involved in many neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and prion disease (Pacelli et al., 2015; Swerdlow, 2018; Wu et al., 2019; Zhang et al., 2020; Wang et al., 2021). SARM1 function has been associated with mitochondrial respiration and mitochondrial dysfunction-induced cell death(Summers et al., 2014; Murata et al., 2018). As we expected, depletion of SARM1 resulted in maintenance of the axonal and mitochondrial morphology following PrP106-126incubation. Next, we determined whether SARM1 was associated with PrP106-126-induced changes in mitochondrial function. We found that downregulation of SARM1 expression limited the effects of PrP106-126on ATP level and MMP. Not surprisingly, overexpression of SARM1 enhanced the PrP106-126-induced loss of ATP and reduction of MMP. Overexpression of SARM1 alone does not always lead to axonal damage or mitochondrial dysfunction, which could also be explained by the autoinhibitory nature of the SARM1 protein. Overexpression of SARM1 is just a condition to increase neuronal vulnerability to experimental processing (Gerdts et al., 2013). In an early study, SARM1 was reported to be involved in induction of axonal degeneration and was associated with intra-axonal mitochondrial dysfunction in the condition of rapid loss of NMNAT2, an enzyme that synthesizes NAD(Loreto et al., 2015). Later studies have explored the NAD+hydrolase activity of SARM1 from different angles (Gerdts et al., 2015; Essuman et al., 2017;Murata et al., 2018; Horsefield et al., 2019; Bratkowski et al., 2020). Our observation of a marked reduction in the abundance of NAD+in PrP106-126-treated N2a cells suggests that NAD+metabolism is imbalanced in ourin vitromodel. Many mutation sites with the potential to regulate the NAD+cleavage activity of SARM1 have been described. The TIR domain of SARM1 is essential for its NADase activity, as mutations E642A and E596K within this domain have been shown to suppress this activity (Essuman et al., 2017; Geisler et al.,2019; Horsefield et al., 2019; Sporny et al., 2020). Ser-548 phosphorylation by JNK is also involved in promoting the NADase activity of SARM1 and switching SARM1 function from promoting mitochondrial transport along axons to inhibiting it (Murata et al., 2018; Xue et al., 2021). We found that PrP106-126failed to induce the loss of NAD+in SARM1(S548A)-expressed N2a cells; the ATP level and MMP were maintained at relatively high levels in comparison with those of the PrP106-126-treated group and SARM1-overexpressed group, and they were not significantly decreased compared with those of the control and SARM1-silenced groups. Therefore, the amino acid substitution at Ser548 suppressed SARM1 NADase activity and limited its further effects on mitochondrial function. These findings illuminate the link between SARM1 activity and mitochondrial dysfunction. Activation of the JNK phosphorylation pathway in prion disease has been previously reported(Carimalo et al., 2005). Further studies of the role of SARM1 in prion disease are warranted. Our finding that NAD+supplementation rescues PrP106-126-induced SARM1-dependent axonal degeneration is in accordance with a previous study that usedin vivoprion models (Wang et al., 2015). In addition,NAD+replenishment prevented mitochondrial fragmentation and dysfunction in PrP106-126-treated N2a cells.
As shown in previous studies, depletion of SARM1 is protective in different disease models, including injury-induced axonal death (traumatic axonal injury or brain injury), chemotherapy-induced peripheral neuropathy,mitochondrial dysfunction-induced cell death of sensory neurons, and photoreceptor cell death and retinal degeneration in Leber congenial amaurosis type nine (Osterloh et al., 2012; Summers et al., 2014; Geisler et al., 2016; Henninger et al., 2016; Marion et al., 2019; Sasaki et al., 2020)In our study, we found that SARM1 silencing and dysfunctional mutation rescued PrP106-126-induced apoptosis and increased cell survival, while SARM1 overexpression aggravated cell death. However, previous studies have found more muted effects of SARM1 deficiency. In one, SARM1 knockout protected against axonal degeneration only in the early stage of experimental allergic encephalomyelitis, and loss of SARM1 did not promote zebrafish axon resealing nor provide motor neuron protection in an amyotrophic lateral sclerosis animal model (Tian and López-Schier, 2020). Another recent study that used a scrapie mouse model reported that SARM1 deficiency failed to change the prion-induced lesion pattern and did not alter PrPScaccumulation;instead, SARM1 deficiency upregulated XAF1 and promoted neuronal apoptosis (Zhu et al., 2019). The above findings indicate that the effect of SARM1 deficiency on axons is protective but not restorative and that SARM1 is involved in the early stage of axonal degeneration.
PrP106-126peptide used as a tractable tool of prion disease study may have limitations. Though PrP106-126peptide recapitulated biological features of PrPSc, but was not infectious. There might be unknown differences in the cell response between the peptide and real prion strain. The effectiveness on the basis of studies with PrP106-126can gain disappointing results in clinical trial. It is a very first step to reveal the role of SARM1 NAD+hydrolase activity in prion disease and the utilization of a new therapeutic strategy. More studyin vivowith real strains is needed in our future work to confirm our findings.
In summary, our findings demonstrate that prion-induced neuronal damage is partially dependent on the NADase activity of SARM1. We showed that depletion of SARM1, dysfunctional mutation of SARM1, and replenishment of NAD+protect against axonal degeneration and mitochondrial dysfunction and promote cell survival in anin vitromodel. Considering that the JNK-c-Jun pathway is activated in prion disease (Carimalo et al., 2005), SARM1 phosphorylation at S548 activates NADase function in injury-induced axonal degeneration (Murata et al., 2018), and SARM1(S548A) is an invalid mutation in PrP106-126-induced NAD+depletion, JNK-SARM1 phosphorylation might be involved in prion disease pathogenesis. The next step is to investigate the mechanisms of SARM1 activation and involvement in prion disease. SARM1 may represent a potential therapeutic target in prion disease.
Author contributions:Study conceptualization, funding acquisition,manuscript review and editing: LFY; experimental implementation, statistical analysis and drafting manuscript: MYL; methodology development: WW, XXZ,JL, ZPL; technical support: WL, ZXS, DMY; data collection: MYL; data analyses:MYL, JL, ZL, MYZ, DDW; manuscript writing guidance and revision: DMZ, XMZ.All authors approved the final manuscript before submission for publication.
Conflicts of interest:The authors declare that they have no conflict of interest.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Guadalupe Álvarez-Hernán, Universidad de Extremadura, Spain.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?