
iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
2022-03-09 07:22JasonHYYeungHenryWaldvogelRichardLMFaullAndreaKwakowsky
中国神经再生研究(英文版) 2022年10期
Jason HY Yeung, Henry J Waldvogel, Richard LM Faull, Andrea Kwakowsky
Alzheimer’s disease (AD) constitutes the largest proportion of dementia worldwide,with a significant associated medical burden.The major pathological hallmarks of AD include the gradual accumulation and deposition of amyloid-beta (Aβ) plaques and hyperphosphorylated tau protein (Revett et al., 2013). Whilst investigations centered around the tau and Aβ hypothesis have been the main focus for the previous decades, the lack of therapeutic solutions has pushed for research into other potential therapeutic targets. Pathological alterations in the glutamatergic system have been postulated to play a central role in the pathogenesis of AD, with intimate downstream and upstream interactions with both Aβ and tau protein(Revett et al., 2013; Yeung et al., 2020a, b;Kwakowsky et al., 2021). Aβ1-42toxicity is mediated in part by N-methyl-D-aspartate receptor (NMDAR) overactivation resulting in elevated intracellular calcium (Ca2+) and subsequent enzyme-induced neuronal death, whilst NMDAR activation has been shown to increase hyper-phosphorylation of neurofibrillary tau (Revett et al., 2013). The complex interrelationship between Aβ1-42,neurofibrillary tau, and the glutamatergic system is still an area of continuing development. A significant disruption of glutamatergic neurons has been well documented in AD, and the subsequent excitatory-inhibitory balance disturbance can potentially contribute to the memory and learning deficits that are characteristic of this condition (Revett et al., 2013; Jurado, 2018;Yeung et al., 2020a, b, 2021; Babaei, 2021;Kwakowsky et al., 2021).
Glutamate mediates the majority of excitatory neurotransmission within the central nervous system, primarily through ligand-gated ionotropic glutamate receptors (iGluRs). These iGluRs are further categorized into three subgroups: the alphaamino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor (AMPAR), NMDAR,and kainate (GluK) receptor classes(Kwakowsky et al., 2021). Each exhibits differences in functionality, with complex interactions between each class. In addition,metabotropic glutamate receptors (mGluRs)mediate a variety of cellular processes,notably fine control over iGluR activity via G protein-coupled or independent cell signaling. mGluRs are also separated into three subgroups; Group I are coupled with phospholipase C, while groups II and III are coupled with adenylyl cyclase. Both iGluRs and mGluRs are formed by a combination of functional subunits, which are classified as GluN1-3 (NMDARs), GluA1-4 (AMPARs),GluK1-5 (kainate), and mGluR1-5(metabotropic). The composition of each receptor dictates its unique functionality- for example, the GluA2 subunit confers the calcium impermeable character of the AMPAR (Revett et al., 2013; Babaei, 2021).
The glutamatergic system has been extensively implicated in AD. Dysfunction appears to be multifactorial, involving a multitude of processes including Aβ binding to glutamate receptors and tau protein tethering to intrinsic cytoskeletal proteins, both of which have been shown to result in receptor overactivation (Babaei,2021). Internalization of glutamate transporters has also been described, with hypotheses this can lead to accumulation of glutamate within the synaptic cleft and subsequent spilling into extrasynaptic areas(Revett et al., 2013; Figure 1A). A leading hypothesis of glutamatergic dysfunction in AD involves the aberrant activation of extrasynaptic glutamatergic receptors,which are associated with inflammatory processes including astrogliosis, release of proinflammatory cytokines, and subsequent apoptosis (Babaei, 2021).In the normal physiological state, there is a fine balance between extrasynaptic and intrasynaptic receptor activation.In AD, Aβ has been shown to facilitate increased activation of receptors beyond the synaptic area, through enhancement of GluN2B-containing NMDAR calcium flux and reduction in GluN1 production,shifting activity towards extrasynaptic receptors (Babaei, 2021). There are reports of pathological synergistic reinforcement,in which extrasynaptic NMDAR activation facilitates the production of tau and Aβ,the presence of which subsequently results in further extrasynaptic activation through NMDAR dysregulation (Figure 1A).Glutamatergic dysfunction in AD is further evident through the therapeutic benefit seen with memantine use. Memantine functions primarily as an NMDAR antagonist,and has the United States Food and Drug Administration approval for the treatment of AD, the only drug approved that is not an anticholinesterase inhibitor or targeting Aβ (Revett et al., 2013). This supports the growing consensus of neuronal toxicity secondary, at least in part, to dysfunction in glutamatergic neurotransmission. The improvement however remains modest at most, indicating that whilst NMDAR overactivity may be a causative factor, it is by no means a breakthrough answer with significant disease-modifying outcomes for AD. More fine-tuned alterations in glutamatergic activity through targeting of subunits may offer a more nuanced method of combating this dysfunction. However,for this, a basic understanding of subunit expression alterations in AD will be required.Despite evidence of glutamatergic system alterations in AD, there has been little insight into specific glutamate receptor subunit alterations in AD. Alterations in subunit composition can have a significant effect on receptor functioning, with downstream implications such as cellular excitotoxicity,oxidative stress, and neuronal cell death(Jurado, 2018). Previous studies performed to quantify glutamatergic changes in the AD brain have relied on older techniques such as autoradiography, or involve the measurement of proxies of subunit expression, such as expression of mRNA transcript levels (Revett et al., 2013). Since initial studies performed in the early 2000s,there has been a lack of progress in the literature surrounding observations regarding the expression patterns and quantitative density changes of glutamatergic subunits in the human AD brain.
Medial temporal lobe atrophy is one of the first changes observed in the brains of AD patients and has the strongest association with the disease besides the most common neuropathological hallmarks. In our recent studies, we attempted to quantify the expression of particular NMDAR and AMPAR subunits within the medial temporal lobe of the AD brain compared to normal control tissue. In particular, areas were separated spatially and analyzed independently,including the CA1, CA2, CA3, and dentate gyrus (DG) of the hippocampus, subiculum,entorhinal cortex, and superior temporal gyrus, to determine any potential regional differences (Yeung et al., 2021). Within the CA1, CA2, CA3, and DG, layer-specific analysis was conducted. From this, we demonstrated quantitative alterations in a variety of glutamatergic receptor subunits(Figure 1B). Quantification of GluA2 subunit expression in human post-mortem hippocampus revealed a significant increase in the stratum (str.) moleculare of the DG in AD compared with control. Increased GluN1 receptor subunit expression was found in the str. moleculare and hilus of the DG, str. oriens of the CA2 and CA3, str. pyramidale of the CA2, and str. radiatum of the CA1, CA2, and CA3 subregions and the entorhinal cortex.GluN2A subunit expression was significantly increased in AD compared with control in the str. oriens, str. pyramidale, and str. radiatum of the CA1 subregion (Yeung et al., 2021).Whilst there have been few human studies previously attempting to elucidate receptor subunit alterations in AD, techniques used such as western blotting are unable to finely analyze specific hippocampal subregions, cell layers and cell types. Utilizing fluorescent immunohistochemistry in combination with confocal microscopy allowed us to quantitatively analyze receptor density in a subregion-, layer and cell type-specific manner. This has revealed interesting,localized alterations that may represent heterogeneous remodeling not previously observed.
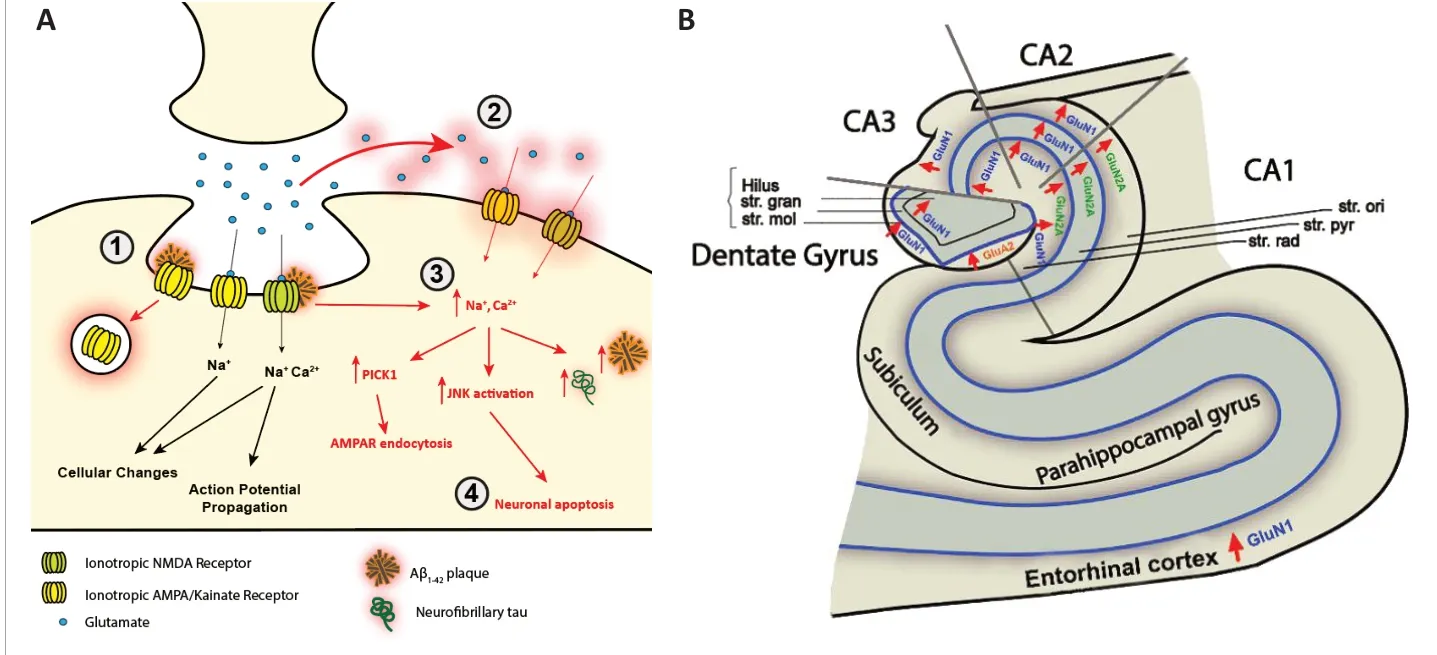
Figure 1 |Pathological alterations of ionotropic glutamate receptor expression in Alzheimer’s disease.(A) AMPAR and NMDAR dysregulation in AD. (1) Aβ induces AMPAR endocytosis and overactivation of NMDARs, resulting in (2) overflow of glutamate into extrasynaptic areas and activation of extrasynaptic GluN2B-containing NMDARs. (3)Aberrant activation of both synaptic and extra-synaptic NMDARs results in the excessive influx of Ca2+ which induces a variety of pathological mechanisms including PICK1-mediated AMPAR endocytosis, increased Aβ and tau production,and increased JNK activation subsequently resulting in (4) neuronal death. (B) Ionotropic glutamatergic receptor subunit alterations in Alzheimer’s disease. Region- and layer-specific expression changes within the hippocampus and entorhinal cortex of the glutamatergic receptor subunits GluA1, GluN1, and GluN2A in AD, suggesting complex spatially dependent expression. AD: Alzheimer’s disease; AMPAR: alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; CA:cornu ammonis; GluA1: glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor type subunit A1;GluN1: glutamate N-methyl-D-aspartate receptor type subunit 1; GluN2A: glutamate N-methyl-D-aspartate type subunit 2A; JNK: c-Jun N-terminal kinase; NMDA: N-methyl-D-aspartate; PICK1: protein interacting with C kinase 1; str. gran:stratum granulosum; str. mol: stratum moleculare; str. ori: stratum oriens; str. pyr: startum pyramidale; str. rad: stratum radiatum.
The AMPAR plays a vital role in triggering the initial cation reflux required for subsequent activation of NMDARs. Previous studies have shown a global downregulation of AMPARs in AD, likely due to Aβ1-42-induced internalization and facilitated receptor degradation (Jurado, 2018). There has also been substantial evidence of tau-mediated alterations in AMPARs in cultured neurons exposed to tau proteins and a variety of transgenic tau mouse models, including htau, rTg4510, and TauRD transgenic mice(Jurado, 2018). The configuration of AMPARs has also been demonstrated to change over time and in response to synaptic plasticity.As mentioned previously, the GluA2 subunit confers a unique characteristic to the AMPAR,resulting in calcium impermeability. GluA2-lacking AMPARs have shown importance in initial long-term potentiation stabilization before subsequently being maintained by a switch from GluA1-containing to GluA2-containing receptors (Jurado, 2018). As such,the composition of AMPARs appears to play a distinct role in processes involved in memory formation. In considering studies supporting the idea of glutamatergic excitotoxicity, an increase in GluA2 subunit expression may demonstrate a protective mechanism against increased cation influx, thereby attempting to inhibit further downstream NMDAR activation. Our study observed an increase in GluA2 subunit expression within the DG str.moleculare (Yeung et al., 2021). Increases in GluA2 subunit expression within the DG is in line with a previous study, which also noted comparatively robust immunolabeling in the DG when compared to other regions, such as the CA1 and subiculum(Armstrong and Ikonomovic, 1996). These heterogeneous changes appear to reflect neuronal loss within vulnerable regions of the hippocampus, whilst the DG is relatively spared in AD. The correlation between GluA2 subunit density and hippocampal regional vulnerability introduces an interesting question as to whether GluA2 upregulation protects against neuronal loss. The mechanism through which these alterations occur remains unknown, although several molecules have been identified to potentially play a role. The protein interacting with C kinase 1 is an integral protein associated with the endocytosis of GluA2-containing AMPARs(Yeung et al., 2021). As protein interacting with C kinase 1 is activated through calciuminduced modification, there is a possibility pathological NMDAR-dependent release of calcium in AD is involved with the GluA2 expression alterations observed within different hippocampal regions.
The NMDARs have been a central focus of AD glutamatergic research. This is primarily due to its potential direct involvement in neuronal excitotoxicity, and also welldocumented interactions with both Aβ1-42and tau. The ability of memantine,an NMDAR antagonist, to produce some,albeit mild, improvement to cognitive and mood symptoms in moderate to severe AD demonstrates its clear involvement in the disease process (Revett et al., 2013).
The GluN1 subunit is ubiquitous within all NMDARs, as it is required for receptor functionality. Thus, an increase in GluN1 subunit expression may imply an aberrant increase in NMDARs, further increasing the excitotoxic effects of glutamatergic overactivity. We observed an increased expression of GluN1 subunit in multiple regions of the hippocampus (Yeung et al.,2021). Whilst mRNA and protein studies of GluN1 have been controversial, our findings are in line with our previous mouse studies showing increased expression in the hippocampus following injection of Aβ1-42(Yeung et al., 2020a). The GluN1 subunit contains the glycine binding site, which acts as a co-agonist and is required for the activation of NMDARs. As GluN1 is an obligatory subunit of all NMDARs, it has not been a central topic for research, with more focus being placed on the more dynamic GluN2/GluN3 subunits. Emerging research,however, has shown some neuroprotective properties of glycine. The application of glycine ameliorated memory impairment and synaptic dysfunction in a D-galactose aging mouse model through downregulation of c-Jun N-terminal kinase (Ullah et al., 2020).Activation of the c-Jun N-terminal kinase signaling pathway has been implicated in apoptotic processes in AD and has also been shown to regulate NMDAR-dependent glutamate release (Nistico et al., 2015). The impact of glycine on modulating NMDAR activity, potentially through interactions with the GluN1 subunit, presents an interesting avenue to pursue.
The GluN2 subunit is further separated into two subtypes, GluN2A and GluN2B,the expression of which changes with brain maturation. An age-dependent reduction in GluN2B expression and concurrent increase in GluN2A has been characterized in both mouse neocortical tissue and human cortical neurons (Pegasiou et al., 2020).These subunits also appear to play opposing roles in AD. GluN2B has been shown to be the target of Aβ1-42-mediated NMDAR dysfunction and Ca2+dysregulation, with prevention of Aβ1-42binding when GluN2B was blocked, whilst antagonism of GluN2A appears to enhance these pathological effects (Babaei, 2021). We reported increased GluN2A subunit expression within the hippocampal CA1 region in AD (Yeung et al., 2021). Other studies examining GluN2A involved homogenates of the entire hippocampus, which masks more localized regional changes. It is unknown whether increased GluN2A subunit expression translates into increased GluN2A-containing NMDAR-mediated neuroprotective effects.The relationship between GluN2A-containing NMDARs and Aβ1-42and tau are extremely complex, with certain molecules implicated in normal GluN2A-containing NMDAR functioning also playing a role in facilitating Aβ1-42toxicity (Yeung et al., 2020a, b; Babaei,2021). However, recent research focusing on specifically targeting GluN2A-containing NMDARs using a novel positive allosteric modulator appears to have therapeutic benefit in neurodegenerative mouse models,indicating the potential neuroprotective role of this subunit (Hanson et al., 2020).The development of positive allosteric modulators offers a promising new approach to targeting the glutamatergic system through specific subunit interactions and represents an exciting developing area of AD research (Babaei, 2021).
We have previously characterized expression changes of glutamatergic receptor subunits and transporters within the CA1, CA3, and DG of the mouse hippocampus, at 3 and 30 days after Aβ1-42injection (Yeung et al.,2020a, b). These studies offered an initial indication of the potential effects of Aβ1-42on the glutamatergic system and demonstrated a relatively robust system against Aβ1-42induced neurotoxic changes particularly within the acute phases of exposure. Our human studies have demonstrated that there are significant glutamate receptor subunit alterations within the AD hippocampus that were not fully replicated by the Aβ1-42injection, indicating more complex physiological processes at play. It is also important to acknowledge the proteolytic process that occurs in post-mortem tissue,thus an examination of this tissue may not be fully replicative of the neuropathological state in its active disease form (Li et al.,2012).
Overall, these findings demonstrate regionaland layer-specific changes of notable glutamatergic receptor subunits in the AD hippocampus and indicate significant remodeling of the glutamatergic system.Not only are changes seen at a receptor expression level, but subtle localized changes to glutamate receptor subunit composition have the potential to lead to significant physiological consequences. We have demonstrated significant alterations in specific iGluR subunits GluA2, GluN1,and GluN2A, each of which has significant impacts on receptor functioning, and has complex interactions with wider signaling pathways. Whether these changes are due to an underlying compensatory mechanism or contribute to further deterioration of neuronal integrity is yet to be determined.Changes in the glutamatergic system are not purely a downstream consequence of AD pathology, with demonstrable reciprocity affecting the propagation of Aβ1-42and tau(Revett et al., 2013; Yeung et al., 2020a,b; Kwakowsky et al., 2021). Both human and mouse model studies demonstrate the complexity of the interaction between AD and the glutamatergic system, which highlights an area that requires further attention in order to better understand the processes behind these molecular alterations. Observed changes should serve as the basis for future work to understand the mechanisms underlying this remodeling.Whilst these studies are promising in demonstrating glutamatergic alteration in AD,there is still a lack of understanding regarding potential functional changes. It is unclear as to why these expression changes occur, and also why certain areas of the hippocampus are affected more than others. Furthermore,the physiological effects of these subunit expression changes are yet to be elucidated.The lack of effective therapies indicates more precise targets, aimed at modulating particular aspects of the glutamatergic system rather than a generalized approach,may be required. We are hopeful that through a better understanding of the drivers and consequences of localized glutamatergic subunit changes, we will be better positioned to identify potential therapeutic targets for the prevention of AD pathology and cognitive decline.
Jason HY Yeung, Henry J Waldvogel,Richard LM Faull, Andrea Kwakowsky*Centre for Brain Research, Department of Anatomy and Medical Imaging, Faculty of Medical and Health Sciences, University of Auckland, Auckland,New Zealand
*Correspondence to: Andrea Kwakowsky, PhD,a.kwakowsky@auckland.ac.nz.https://orcid.org/0000-0002-3801-4956(Andrea Kwakowsky)
Date of submission: August 29, 2021
Date of decision: October 18, 2021
Date of acceptance: November 13, 2021
Date of web publication: February 28, 2022
https://doi.org/10.4103/1673-5374.335804
How to cite this article:Yeung JHY, Waldvogel HJ,Faull RLM, Kwakowsky A (2022) iGluR expression in the hippocampal formation, entorhinal cortex, and superior temporal gyrus in Alzheimer’s disease.Neural Regen Res 17(10):2197-2199.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long asappropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Arturo Ortega, CINVESTAV Departamento de Toxicologia, Mexico.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?
- Novel insights into the pathogenesis of tendon injury:mechanotransduction and neuroplasticity